| Название файла/программы | Команда | Результат | Описание |
| Исходный файл | Ссылка для скачивания | - | |
| 1. Анализ качества чтений программа FastQC | установлена на компьютер, на вход получает файл .fastq | chr2_fastqc.html | |
| 2. Очистка чтений программа Trimmomatic | Удалить буквы с качеством ниже 28 с конца
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr2_.fastq chr2_1.fastq TRAILING:28 Удалить последовательности длиной меньше 50 п.о. java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr2_1.fastq chr2_2.fastq MINLEN:50 | chr2_2.fastq | Команда TRAILING удаляет буквы с качеством ниже установленного с конца рида. Команда MINLEN удаляет риды длиной меньше установленного значения |
| Сравнение качества полученного файла с первоначальным программа FastQC | установлена на компьютер, на вход получает файл .fastq | -chr2_2_fastqc.html | |
| 3.Картирование чтений Путь до файлов, содержащих программу Hisat2 export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
Индексирование референсного файла: hisat2-build chr2.fasta chr2
Построение выравниваний референса и прочтений: hisat2 -x /nfs/srv/databases/ngs/azbukinanadezda/chr2 -U /nfs/srv/databases/ngs/azbukinanadezda/chr2.fastq --no-softclip --no-spliced-alignment >chr2.sam | /nfs/srv/databases/ngs/azbukinanadezda/chr2.sam | Команда hisat2-build индексирует референсную последовательность ДНК. На выход выдает 6 файлов (*.1.ht2, *.2.ht2 и т.д.). Все они нужны для выравнивания сиквенсов с референсов. Интересно заметить, что исходный fasta файл с референсом больше не нужен. В общем, эта команда способна индексировать геномы любого размера. Если размер референса меньше 4*10^9 п.н., то индекс записывается в 32-битном формате ("small"), если больше - то в 64-битном и выходные файлы тогда называются *.1.ht21,"large". Пользователь может изменить выдачу с помощью флага --large-index.
Команда hisat2 выравнивает прочтения и референс. Флаг -x указывает на путь к проиндексированному референсу, флаг -U - к файлу с ридами. Флаг --no-softclip запрещает подрезать нуклеотиды с концов ридов для выравнивания, --no-spliced-alignment запрещает разрезать рид и искать его куски на экзонах (тк мы секвенируем ДНК, а не мРНК). |
| 4. Анализ выравнивания пакет samtools |
Переведение в бинарный формат : samtools view -bS chr2.sam > chr2.bam
Сортировка по координате в референсе начала чтения: samtools sort chr2.bam chr2sorted Индексация файла samtools index chr2sorted.bam | /nfs/srv/databases/ngs/azbukinanadezda/chr2sorted.bam | samtools view переводит файл с выравниваниями в бинарный формат. samtools sort сортирует полученный бинарный файл по начальным координатам (по умолчанию). Затем файл индексируется. | 5. Поиск SNP. Создание файла с полиморфизмами в формате .bcf | samtools mpileup -I chr2sorted.bam -uf chr2.fasta -g -o task5 | /nfs/srv/databases/ngs/azbukinanadezda/task5 | Файл со списком отличий | bcftools call task5 -cv >chr2.vcf | chr2.vcf |
| 6. Анализ Annovar | Подгодовка файла perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr2.vcf > chr2.avinput | /nfs/srv/databases/ngs/azbukinanadezda/chr2.avinput | |
| Поиск rs, dbSNP | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs -build hg19 -dbtype snp138 chr2.avinput /nfs/srv/databases/annovar/humandb/ | Здесь и далее все команды организованны по принципу perl /скрипт/ - запускает скрипт,-build указывается версия сборки генома человека, -dbtype в какой базе данных искать, и в конце файл на вход. | |
| Refgen | perl /nfs/srv/databases/annovar/annotate_variation.pl -out refgene -build hg19 chr2.avinput /nfs/srv/databases/annovar/humandb/ | ||
| 1000genomes | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out ex1 chr2.avinput /nfs/srv/databases/annovar/humandb/ | ||
| Gwas | perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog chr2.avinput /nfs/srv/databases/annovar/humandb/ | ||
| Clinvar | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 chr2.avinput -outfile clinvar /nfs/srv/databases/annovar/humandb/ |
Очистка чтений и анализ их качества
Описание команд и ссылки на результаты доступны в таблице (все необходимые изображения расположены на html странице, ссылка в таблице). В исходном файле до чистки было 10410 последовательностей длиной 40-100 нуклеотидов. В результате чистки с помощью прорамммы Trimmomatic были удалены нуклеотиды с плохим (ниже Q=28, Q=-10lg(вероятности ошибки)) качеством, а также риды короче 50 нуклеотидов. Поэтому на картинке качества сиквенсов "пропали" риды с длинными усами, а коэффициент эксцесса пика распределения качества ридов увеличится, он станет более острым. В результате файл стал содержать 10191 последовательностей. Однако в обоих ридах стоит флажок "failure" по критерию содержания GC на последовательность (ссылки на результаты в виде html страниц смотри в таблице). Этот флажок загорается, если имеются отклонения от нормального распределения содержания GC-пар в более чем 30% ридов. В распределениях, полученных на основе данных моих файлов (ссылка в таблице), имеется сдвиг влево, что является индикатором автоматической ошибки, не зависящей от позиции основания. Кроме того на левой половине пика имеется много остроконечных локальных максимумов, которые служат индикатором проблем с библиотекой. Обычно это обозначает наличие специфического загрязнения, например димеры адаптеров. Но в наших файлах адаптеры уже должны были быть удалены (это видно в поле Adapter Content), поэтому сложно говорить о причинах таких сдвигов. Другое поле, содержащее предупредительные сигналы - распределение длины сиквенсов. В моих файлах пости все последовательности превышают в длину 97 нуклеотидов, однако некоторые содержат меньшую длину. В описании программы указано, что индикаторы из этого раздела можно игнорировать, так как для некоторых секвенаторов наличие ридов неодинаковой длины является нормой. По остальным критериям и изначалный и конечный файл удовлетворяют параметрам качества.Другими интересными полями выдачи являются проверка распределения нуклеотидов по сиквенсам. В норме все 4 линии должны быть параллельны друг другу и разница в содержании между А и Т и между G и С не должна превышать 10 процентов (правило Чаргоффа). Еще есть поле, описывающее содержание N-неопознанного нуклеотида в ридах. Оно показывает качество секвенирования, насколько прибор смог различить последовательность нуклеотидов. Другим немаловажным показателям качеста секвенирвоания является количество дуплицированных сиквенсов. Маленький уровень дупликация является индикатором высокого уровня покрытия таргетной последовательности, а высокий - ошибки в проведении ПЦР (что почему-то каких-то последовательностей сильно больше, чем других).
Картирование чтений и анализ выравнивания
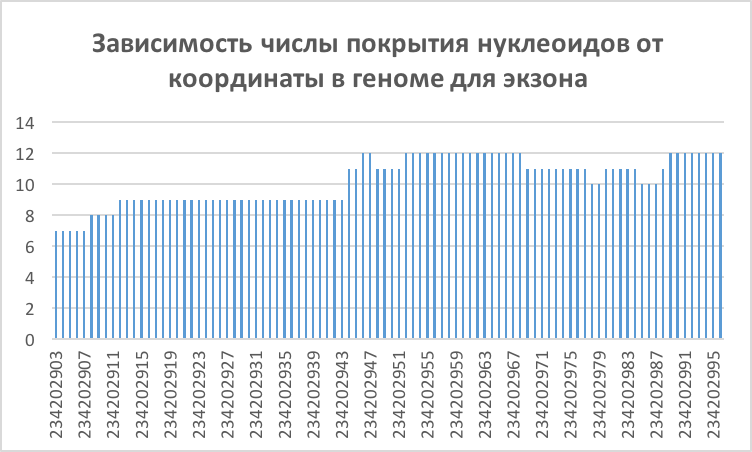
Используемые команды и программы указаны в таблице 1. Очищенный файл содержал 10191 ридов, из них с помощью программы Hisat2 все были непарными, 10140 (99.50%) выровнены ровно 1 раз, 4 (0.04%) выровнены больше 1 раза и 47 (0.46%) вообще не удалось картировать на хромосому.С помощью команды samtools depth chr2sorted.bam >depth.tsv я получила табличку с информацией о том, сколько раз был покрыт каждый нуклеотид. C помощью программы excel я отсортировала данные, выбрала нуклеотид, который покрыт много раз (234204021). Ссылка на табличку. Затем с помощью программы genome brouser я определила положение этого нуклеотида на хромосоме. Он попал на ген ATG16L1 . Файл, содержащий экзоны этого гена (в том числе 3' и 5 ' нетранслируемые экзоны доступен по ссылке в формтае .fasta. Для примера был взят экзон chr2:234202903-234202996. еще раз была запущена комнада samtools depth chr2sorted.bam -r chr2:234202903-234202996 >depth2.tsv. Среднее число покрытий 10,21, медиана -11,мода -9 (расчеты в том же файле excel). Также была построена гистограмма распределения глубины покрытия нуклеотида от координаты генома (см рисунок 1). Я думаю, можно утверждать, что покрытие данного экзона довольно равномерное.
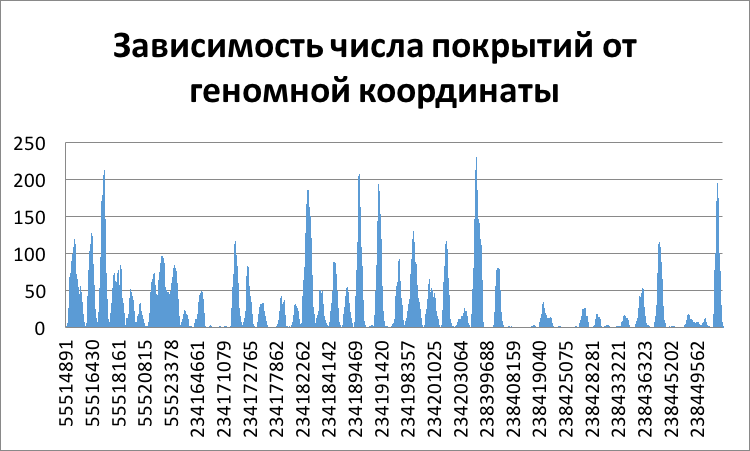
Дальше мне стало интересен общий профиль покрытия всей хромосомы. Среднее покрытие составляло 26,7, мода -1, медиана -9. То есть половина нуклеотидов генома покрыта меньше 9 раз. Если взглянуть на гистограмму распределния покрытий по хромосоме видно, что на протяжении всего геноа чередуются участки с высоким и с низким покрытием (см. рисунок 2).
Анализ SNP
Необходимый команды описаны в таблице 1. Описание полиморфизмов содержится в таблице 2.| Координата | Тип полиморфизма | Референс | Чтение | Глубина покрытия | Качество чтения |
| сhr2:55516588 | Замена | G | C | 23 | 184,999 |
| сhr2:55523309 | Замена | C | A, G | 36 | 211,003 |
| сhr2:238454154 | Замена | C | A | 101 | 225,009 |

Затем я проаннотировала эти SNP в refgene, базе данных, основанной на геномной разметке. Команда указана в таблице. В результате на выход мы получаем три файла: refgene.variant_function содержащий информацию о том, в какое место попал SNP (3' и 5' нетранслируемые области, интроны, экзоны обычных генов, экзоны некодирующих РНК, области, близкие к соединениям спалйсинга, upstream - в пределах 1кб до начала транскрипции, downstream-в пределах 1 кб после сайта окончания транскрипции, интергены), refgene.exonic_variant_function(содержит подробную информацию о том, синонимичная ли замена или нет, какая аминокислота на какую поменялась), rs.log(stdout). В экзоны попало 6, в интроны - 57, ncRNA-exonic -1, UTR3-6, UTR5-2. Гены- ATG16L1,MLPH, CCDC88A, MIR6811. Подробнее о SNP, попавших в экзоны смотри в таблице 3.
| Ген | Координата | АК референс | АК чтение | Нуклеотид референс | Нуклеотид чтение |
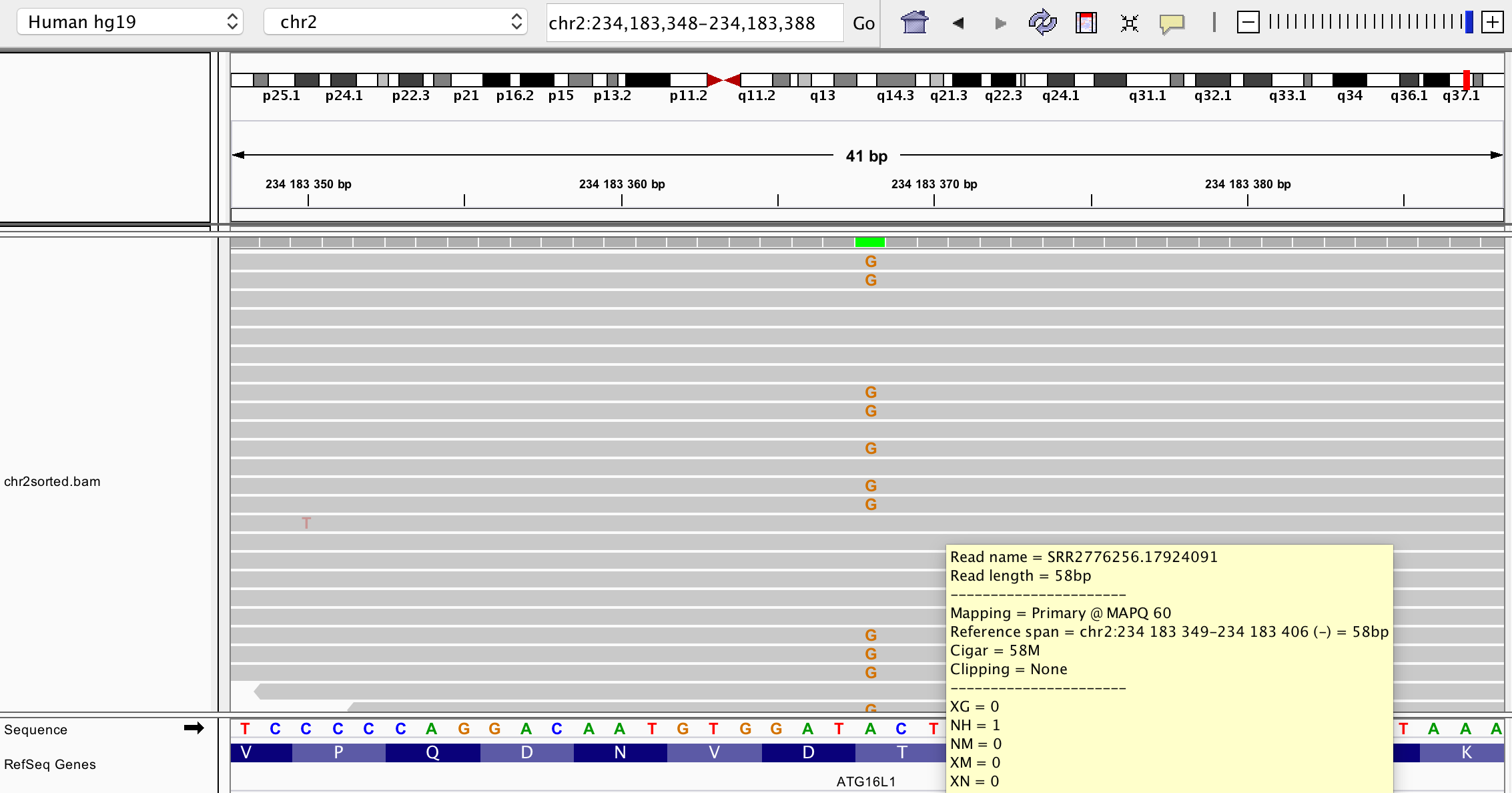
| ATG16L1 | сhr2:234183368 | T | A | A | G |
| MLPH | сhr2:238427194 | L | P | T | C |
| MLPH | сhr2:238427251 | G | D | G | A |
| MLPH | сhr2:238443226 | H | R | A | G |
| MLPH | сhr2:238449007 | V | A | T | C |
| MLPH | сhr2:238449107 | E | E синонимичная замена | A | G |
Затем SNP были проаннотированы с помощью базы данных Gwas, основанная на полногеномном поиске ассоциаций между генотипическими и фенотипическими признаками. Всего нашлось 4 SNP: 55516323 связан с ростом человека, 234173503 и 234183368 с болезнью Крона, а 238443226 с раком простаты.
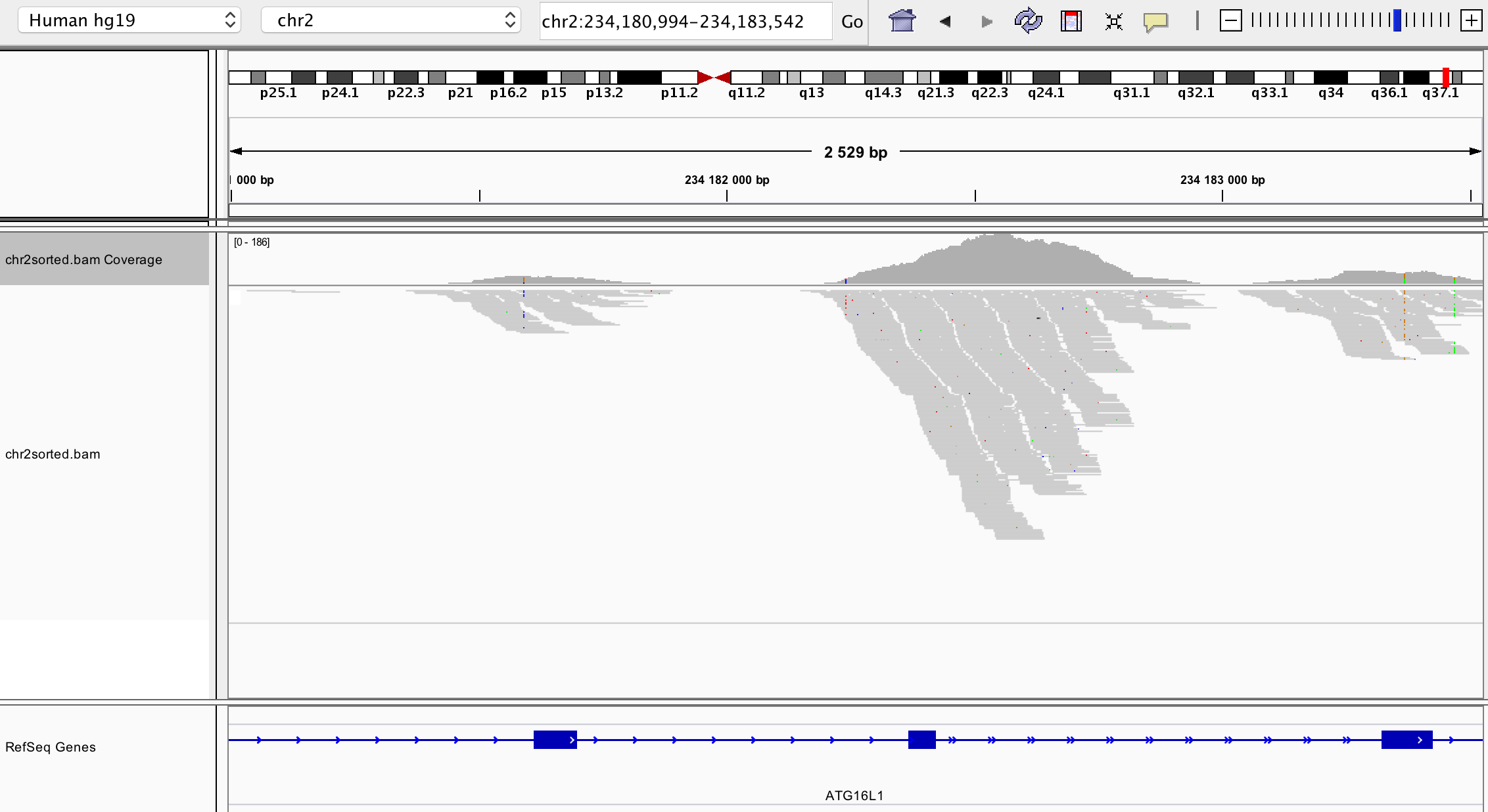
Чтобы определить, имеют ли SNP этого пациента какую-либо клиническую аннотацию, был произведен поиск по базе данных Clinvar, содержащую инфрмацию о генотипической вариабильности и ее связи с человеческим здоровьем. Оказалось, что нуклеотид с номером 234183368 (замена A на G) связан с заменой 300-ой АК треонина на аланин, идентификатор в этой базе данных:RCV000001189.2. , и это приводит к предрасположенности к Inflammatory bowel disease ( воспаление кишечника), к которым как раз и относится болезнь Крона. Чаще всего заболевание развивается до 30 лет [1]. Данная мутация случается в гене ATG16L1 - autophagy related 16 like 1( Gene ID: 55054), белок, закодированный в этом гене входит в состав большого комплекса, ответственного за аутофагию - процесс, направляющий компоненты внутриклеточного матрикса на деградацию в лизосомы [2]. На рисунке 5 представлена визуализация этого полиморфизма с помощью IGV.