| Название файла/программы | Команда | Результат | Описание |
| Исходные файлы | /nfs/srv/databases/ngs/azbukinanadezda/pr14/SRR4240356.fastq | - | |
| 1. Подготовка чтений программой trimmomatic [1] подготовка адаптеров | seqret @mylist.txt adapters.fasta | /nfs/srv/databases/ngs/azbukinanadezda/pr14/adapters.fasta | Файлы с адаптерами были объединены в общий файл. |
| Удалиние остатков адаптеров | java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240356.fastq 1.fastq ILLUMINACLIP:adapters.fasta:2:7:7 | /nfs/srv/databases/ngs/azbukinanadezda/pr14/1.fastq |
Команда ILLUMINACLIP первым параметром использует название fasta -файла, содержащего адаптеры, праймеры для ПЦР и т.д. Второй параметр определяет максимальное число несоответствий, которое допускается при определении соответствия. Третий параметр показывает, насколько аккуратным и точным должно быть соответствие между двумя "соединенными адаптерами" ('adapter ligated' ) ридами должно быть для палиндромного выравнивания ридов. ЧЕтвертый параметр определяет насколько точным должно быть соответствие между любыми адаптерными последовательностями и ридами. |
| Удаление плохих букв с концов чтений | java -jar /usr/share/java/trimmomatic.jar SE -phred33 1.fastq 2.fastq TRAILING:28 MINLEN:30 | /nfs/srv/databases/ngs/azbukinanadezda/pr14/2.fastq | Команда TRAILING удаляет буквы с качеством ниже установленного с конца рида. Команда MINLEN удаляет риды длиной меньше установленного значения. | 2. Подготовка k-меров. | velveth velveth 29 -fastq -short 2.fastq | /nfs/srv/databases/ngs/azbukinanadezda/pr14/velveth На выходе программы записывается три файла:Log, содержащий общую информацию -протокол выполнения, Sequences - содержит пронумерованные последовательности ридов и файл Roadmaps. Два последних необходимы для следующих этапов. | Программа velveth [2] первым параметром принимает имя диретории, куда будут записаны выходные файлы, вторым - длина к-меров, третьим - формат входного файла, четвертым - тип чтений (короткие/длинные/парные/непарные) и в конце пишется название исходного файла. ls |
| Сборка | velvetg velveth25 | /nfs/srv/databases/ngs/azbukinanadezda/pr12/chr2.2intersect.bed или chr2.1intersect.bed | Дополнительные параметры для сборки коротких ридов -cov_cutoff, можно удалять куски с покрытием меньше указанного. Иногда это бывает полезно, а иногда- теряется нужная информация. |
Подготовка чтений программой trimmomatic
Ссылка на исходные данные моего проекта SRR4240356. После подготовки файла с адптерами было проведено удаление остатков адаптеров. Отчет о выполнении доступен здесь. В первоначальном файле было 7511529 ридов, после чистки было удалено 153091 (2,04%), осталось 7358438 (97,96%) ридов.После этого были удалены плохие буквы на концах чтений, а также чтения, с длиной меньше 30. На вход на этом этапе подавалось 7358438 ридов, удалено 73655 (1,00%), осталось 7284783 (99,00%). Исходный файл имел размер 793 999 866 байт, после чистки - 757 103 626 байт. Пройдя по ссылкам, можно увидеть результат выдачи программы fatqc (команда factqc 'имя файла'), для первоначального варианта и конечного. Можно заметить, что действительно были удалены риды, с рамахом неудовлетворительного (ниже 28) качества, а также удалены overrepresented sequences, то есть действительно адаптеры были удалены. Интересен тот файт, что содержание пар G-C составляет примерно 30%, а A-T -70%, то есть исследуемая бактерия не термофильна (поле per base sequence content. Причем видно, что в первоначальном файле содержание колеблется от рида к риду, а в очищенном- равномерное по всем ридам).
Подготовка k-меров и сборка
С помощью программы velveth были подготовлены k-меры нужной длины. Затем была проведена сборка программой velvetg[2], работающий на основе графов де Брайна для k-меров длины 25 и 29. Так как командой sort отсортировать данные не удалось, они были обработаны с помощью excel. Основные параметры приведены в таблице 2.| Параметр | k-mer length=25 | k-mer length=29 |
| N50 | 5 408 | 73 133 |
| Общая длина, п.н. | 695 924 | 664 866 |
| Число контигов | 3 319 | 608 |
| Самые длинные контиги | a)ID:51, lgth 14 084, cov 80.41;
b)ID:13,lgth 13 372, cov 81.19; c)ID:59,lgth 13 359, cov 76.87. | a)ID:7, lgth 115 468, cov 51.99 ; b)ID:19, lgth 106 076, cov 45.77; c)ID:8, lgth 75 082, cov 54.26. |
Нашлись контиги с аномальным покрытием. Медиана покрытия составила 10,82. Контиг с максимальным (312 992) покрытием имеет номер 517 и длину 1 нуклеотид, поэтому понятно, почему у него такое большее покрытие. В файле stats.txt содержится информация о всех 610 узлах построенного графа, но в файле contigs.fa содержатся только поулченные контиги, с минимальной длиной, соответствующей длиной k-mer =29 нуклеотидов (таких контигов 16 штук).
Анализ полученных данных
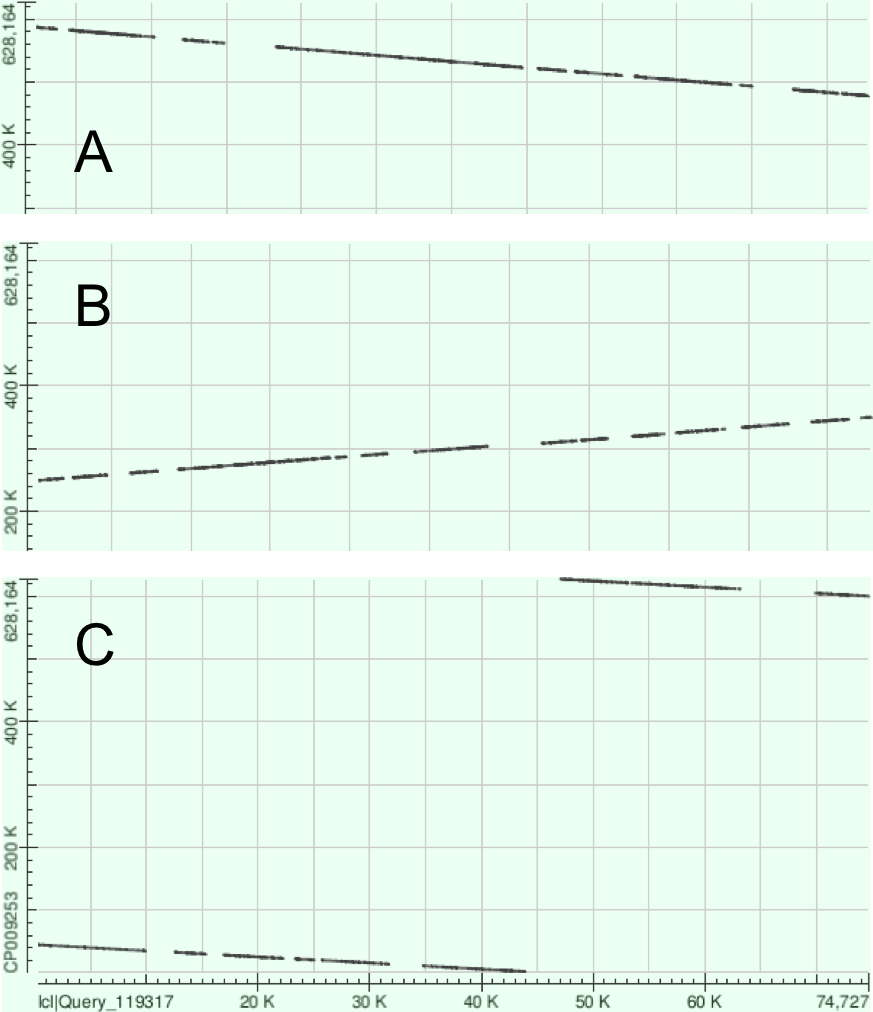
Для анализа собранных чтений были извлечены .fasta файлы самых длинных контигов и сравнены с уже собранной хромосомой Buchnera aphidicola (GenBank/EMBL AC — CP009253) программой blast2seq (megablast). Результаты приведены в таблице 3. Согласно таблице видно, что процент несоответствий (миссматчей) и гэпов в выравниваниях примерно одинаковый между 3 -мя контигами. Расчеты производились в программе excel. Результаты доступны в том же файле, что и для k-меров excel.| Ссылка на fasta, длина выравнивания | E-value | Покрытие | Идентичность | Гэпы | Несоответствия |
| Контиг 7. 86 586 п.н. | 0.0 | 73% | 81% | 2 205 (2,55%) | 16 324 (18,85%) |
| Контиг 19. 73 984 п.н. | 0.0 | 68% | 79% | 1 938 (2,62%) | 14 770 (19,96%) |
| Контиг 8. 55 803 п.н. | 0.0 | 73% | 83% | 1 172 (2,10%) | 10 194 (18,27%) |
| Параметр | Половина ридов | Все риды |
| N50 | 30 234 | 73 133 |
| Общая длина, п.н. | 658 385 | 664 866 |
| Число контигов | 339 | 608 |
| Самые длинные контиги | a)ID11, lgth 42 588, cov 24.60;
b)ID:6, lgth 41 368, cov 24.77; c)ID:8, lgth 36 360, cov 25.97. | a)ID:7, lgth 115 468, cov 51.99 ; b)ID:19, lgth 106 076, cov 45.77; c)ID:8, lgth 75 082, cov 54.26. |
Видно, что геном собран полностью (общая длина примерно одинаковая), однако N50 в два раза меньше и число контигов тоже в два раза меньше (видимо мало коротких, так как значимых - длиной больше 100 п.н. в изначальном -56, а в урезанном - 64). Кроме того, меньше размер самых длинных контигов (раза в 3), а покрытие в среднем ниже в два раза. То есть можно сделать вывод, что геном собрать можно, однако достоверность будет ниже, но для получения чернового варианта такое количество ридов вполне сгодится.
Сборка с помощью SPAdes
Тот же самый файл 2.fastq был собран с помощью другой программы Spades [3]. Команда для запуска spades.py -s 2.fastq -k 29 -o spades. -s -флаг, указывающий, что исходный файл содержит простые риды, -k -длин k-меров, -o -директория, куда будут записаны выходные файлы. В моем случае этап Mismatch Correction был пропущен, так как риды одиночные, не парные, и нет референсных ридов.В общем, программа SPAdes содержит гораздо больше опций для работы с разными типами ридов ( по сравнению с velvet), можно задавать файл с достоверными референсными ридами для сравнения на этапе корректировки и проверки ридов, собирать one-cell sequence (--sc), а также продолжать работу с места ошибки. также стоит отметить, что эта программа прездназначена конкретно для сборки бактериальных геномов. Но зато она работает гораздо дольше (вся процедура около 40 мин), чем velvet.
Выходные файлы содержат: contigs.fasta, contigs.fastg – последовательности контигов в разных форматах (fastg - похож на фаста, только содержит куски, записанные в виде графов, что полезно, не нужно записывать много фаста файлов, например для разных аллелей), scaffolds.fasta, scaffolds.fastg – последовательности скаффолдов, corrected/ – директория, содержащая файлы после коррекции ридов (актуально, если риды спаренные), configs/ – рабочие файлы для коррекции ридов, params.txt –параметры SPAdes в этой сессии, spades.log, dataset.info – общая информация о программе SPAdes.
В таблице 5 приведено сравнение работы программы velvet и SPAdes (статистика для программы SPAdes получена с помощью QUAST [4], информация о конкретных контигах получены из названия контигов в fasta-файле, обработано в excel). Интересно, что значимых контигов (длиной больше 100 п.о. в программе velvet получилось 56, а в SPAdes - 111), относительно общего количества гораздо больше, чем полученных программой velvet и они в среднем длиннее.
| Параметр | SPAdes | Velvet |
| N50 | 13 841 | 73 133 |
| Общая длина, п.н. | 663 444 | 664 866 |
| Число контигов | 143 | 608 |
| Самые длинные контиги | ID:1, lgth 32 944, cov 49,08 ; b)ID:3, lgth 31 521, cov 52,40; c)ID:5, lgth 31 329, cov 48,65. | a)ID:7, lgth 115 468, cov 51.99 ; b)ID:19, lgth 106 076, cov 45.77; c)ID:8, lgth 75 082, cov 54.26. |
Список источников
[1] Программа Trimmomatic.[2] Программа Velveth.
[3] Программа Spades.
[4] Программа QUAST.