Алгоритмы филогенетической реконструкции
Таксоны
Мы строили деревья для птичек из прошлого практикума.
Выравнивания и подготовка файлов
Снчала мы получили последовательности из swissprot по списку AC seqret @cyb.list cyb.fasta . Затем мы сделали выравнвание для списка AC c помощью программы muscle: muscle -align cyb.fasta -output cyb-alignment.fasta
И маленькой программкой на питоне адаптировали формат файлов для программы Fastme:
from Bio import AlignIO
from sys import argv
inh = open("cyb-alignment.fasta", "r")
outh = open("cyb.phy", "w")
alignment = AlignIO.parse(inh, "fasta")
AlignIO.write(alignment, outh, "phylip-relaxed")
inh.close()
outh.close()
Fastme и IQ-Tree
FastME (Fast Minimum Evolution) — это программное обеспечение для филогенетического анализа, которое строит деревья эволюции на основе матриц расстояний между биологическими последовательностями. Он работает, быстро вычисляя расстояния, строя начальное дерево и оптимизируя его топологию с помощью алгоритмов перестановки ветвей для минимизации длины дерева
IQ-TREE основан на методе максимального правдоподобия (ML). Он принимает выровненные последовательности, автоматически подбирает лучшую модель эволюции, строит дерево, используя эвристические алгоритмы поиска, и оценивает его надежность с помощью ультрабыстрого бутстрепа, что обеспечивает высокую скорость и точность.
Следующими командами строили деревья:
fastme -i cyb.phy -pP -o Ptree.from.fastme - параметр p-distance fastme -i cyb.phy -pM -o Mtree.from.fastme - параметр MtREV iqtree -s cyb.phy - программой IQ-Tree
Пару слов про параметры
p-distance это метрика, которая высчитывает попарную меру сходства и различия, то есть он считает долю несовпадающих позиций в двух выровненных последовательностях. То есть вероятность замены последовательных позиций или вероятность замен на одной позиции несколько раз он не учитывает.
mtREV это mitochondrial REVersible. Это модель, которая специально сделана для митохондриальных белков и учитывает вероятности замены одних аминокислот на другие. На основе наблюдений, то есть эмпирически, была создана матрица замен (mtREV для позвоночных), и, используя метод максимального правдоподобия, она помогает оценить степень родства митохондриальных белков.
Все эти алгоритмы, включая IQtree, создают неукоренённые деревья, то есть они показывают только родственные связи между видами, но не определяют, где находится "начало" эволюции для выбранной клады. Чтобы корректней оценивать дерево его необходимо переукоренить.
Сами деревья
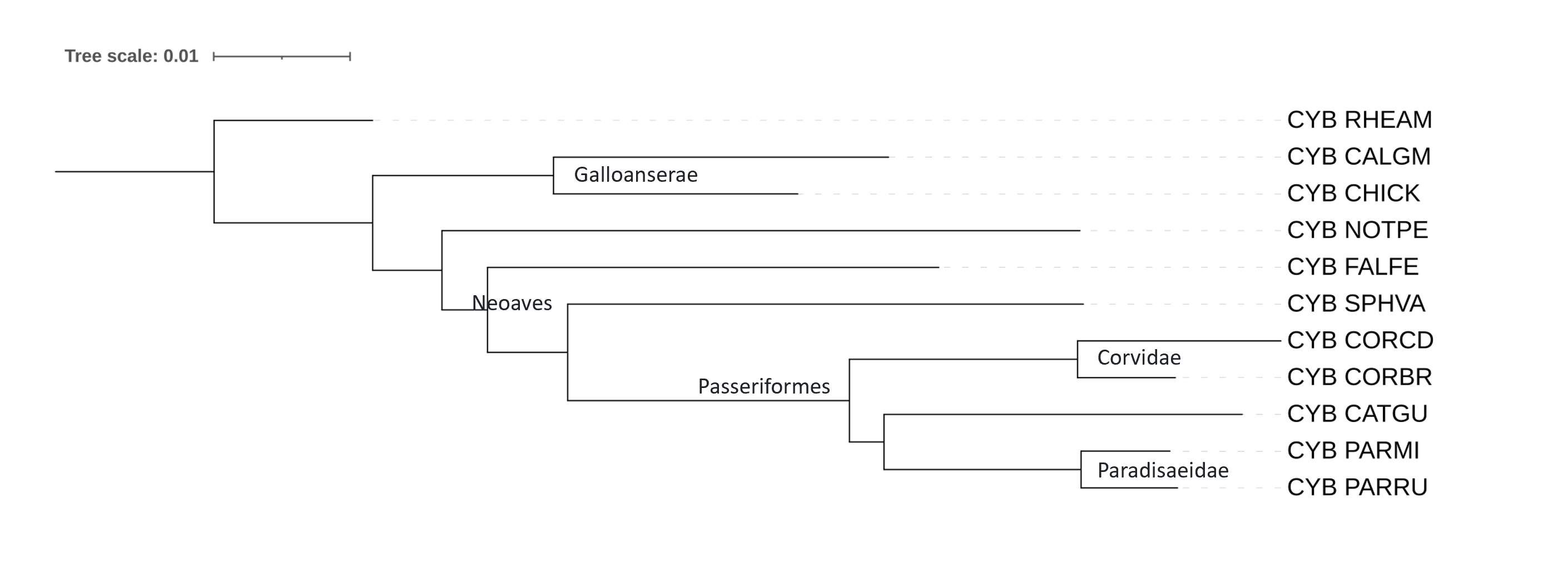
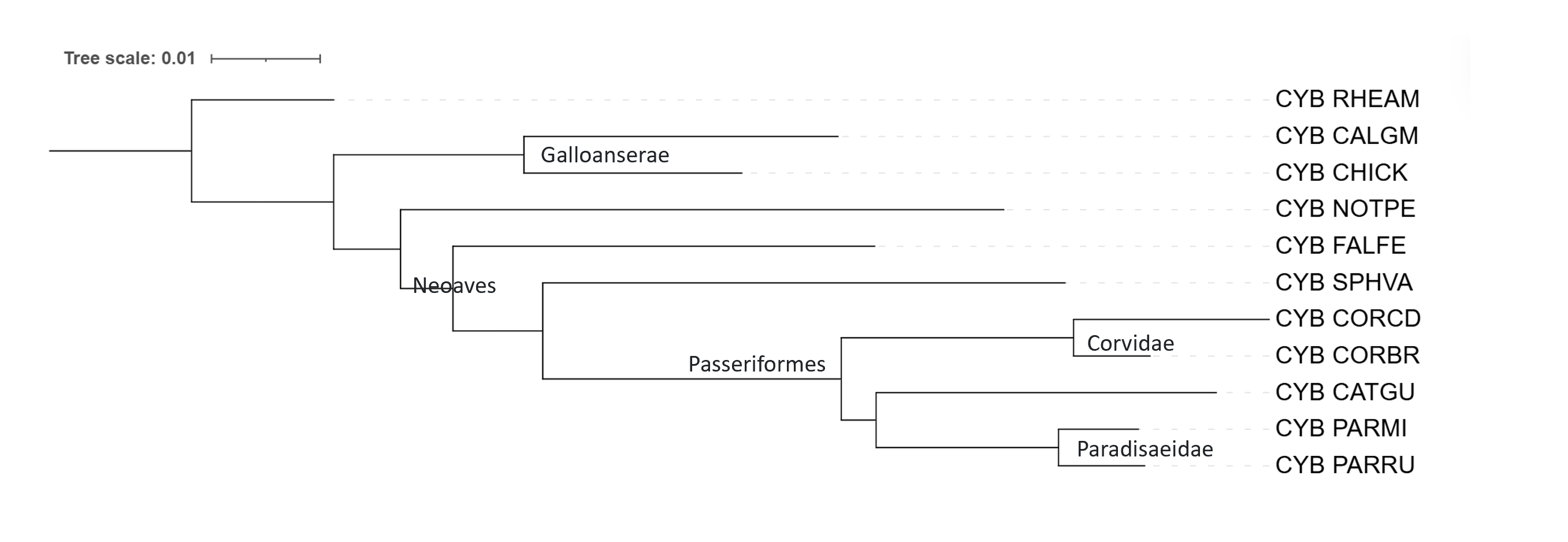
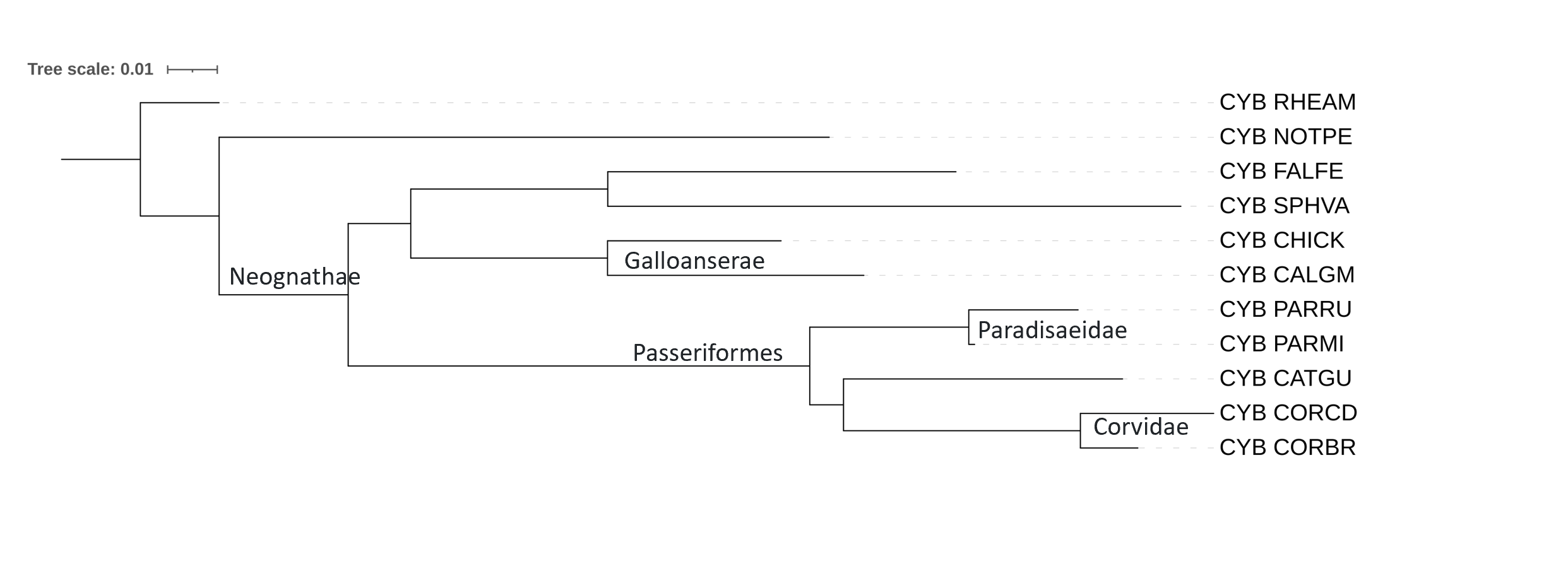
Изначально полученные деревья iTOL неукоренённые (изображения укоренены в случайное место). Чтобы проводить корректное сравнение, корень должен находиться на ветви, которая отделилась ровно один раз и до начала диверсификации изучаемой группы. Самой древней и наиболее рано отделившейся является Rhea americana (RHEAM) — это Обыкновенный нанду, обитатель Южной Америки. Есть хорошие красивые картинки, подтверждающие, что бескилевые птицы (Paleognathae - страусы, нанду и наш Чилийский степной тинаму NOTPE) отделились раньше остальных птичек, которые Neognathae.
Подписаны верно сформированные клады.
Выводы
Два дерева построенные Fastme топологически не отличаются. По числу совпавших групп с таксономическим деревом они такие же, как дерево от IQtree. Но дерево от IQtree мне кажется лучше, так как Fastme неправильно аннотировали NOTPE, он должен быть в одной кладе с RHEAM, они Palaeognathae и это довольно грубое нарушение систематики.
Неправильно указан порядок ветвей с FALFE, SPHVA и далее. SPHVA это Жёлтобрюхий дятел-сосун, он должен быть в кладе Afroaves, в то время как FALFE и воробьинообразные Passeriformes (CORBR, CORCD, PARRU, PARMI, CATGU) это Australaves.
Все трое не справились с кладой воробьинообразных: CATGU должен быть вне клады Corvoidea (к которой принадлежат CORBR, CORCD, PARRU, PARMI), этот дрозд должен быть в своей кладе Turdidae (семейство дроздовые).