Анализ транскриптомов
Задание 1. Оценка качества чтений
| Input | Output | Программа | Что делает |
| chr8.2.fastqc | chr8.2_fastqc.zip | fastqc chr8.2.fastqc |
Получаем возможность проанализировать наши чтения |
| chr8.2.fastq | cleaned.fastq | java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr8.2.fastq cleaned.fastq TRAILING:20 |
Отбираем риды качеством выше 20 |
| cleaned.40.fastq | cleaned.40_fastq.zip | java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 cleaned.fastq cleaned.40.fastq MINLEN:45 |
Отбираем последовательности длины более 45 |
| cleaned.2.fastq | cleaned.2_fastq.zip | fastqc cleaned.2.fastq |
Получаем возможность проанализировать очищенные чтения |
Файл после работы с Trimmomatic
Прочтения сразу были достаточно хорошего качества (минимум 28 у самого "плохого" уса, точки
10%) До чистки по числу bp, длины последовательностей были 39-51, что, по сути, не критично для ридов,
полученных секвенированием РНК
Для улучшения параметра Per tile sequence quality достаточно ограничения в длинах = 45
Тепловая карта Per tile sequence quality До и После ограничения по длинам ридов
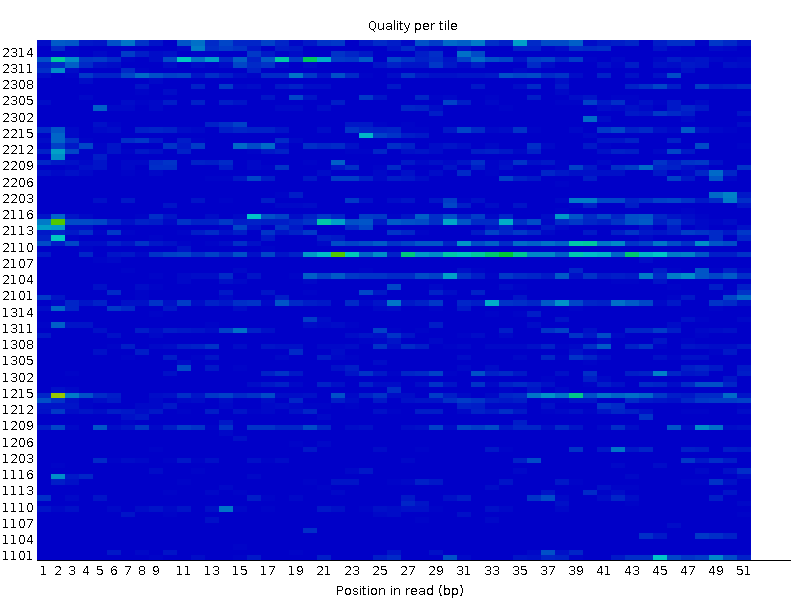 |
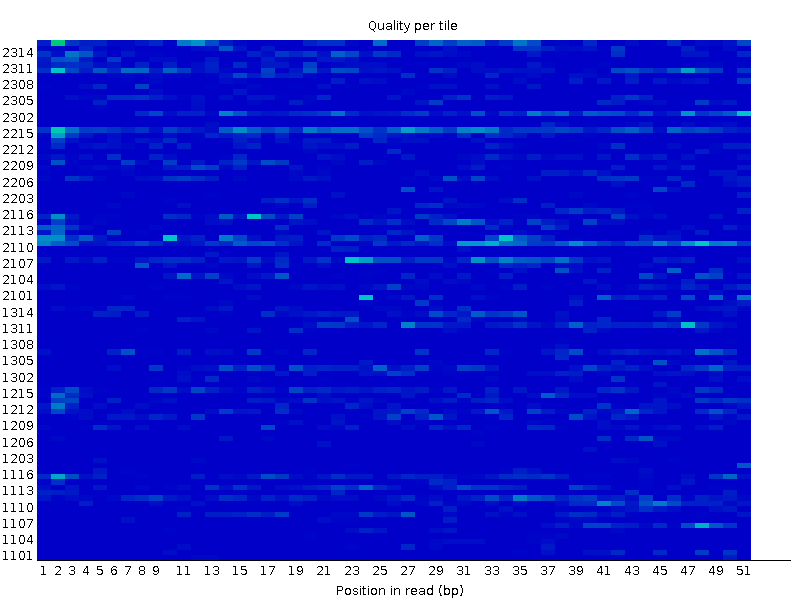 |
Этот график показывает качество нуклеотидов каждого рида; все они характеризуются цветами
различных оттенков - теплого или холодного (холодные цвета показывают качество нуклеотида выше среднего,
теплые, соответственно - ниже)
Таким образом, если взглянуть на теплокарту до чистки ридов по их длине, то мы видим несколько остатков зеленоватого
цвета. После чистки с помощью Trimmomatic все риды окрашены в холодные цвета, а значит, в нашей выборке
остались чтения с нуклеотидами качеством выше среднего
Также, даже после чистки у нас остались "критические" значения некоторых полей - Per base sequence content,
Sequence Duplication Levels и предупредительные отметки параметров Per sequence GC content,
Sequence Length Distribution и Overrepresented sequences
Модуль Per base sequence content выдает ошибку, когда различие между основаниями пары более 20% в определенной позиции,
что особенно явно отмечается в начальных позициях (в нашем случае). Причиной может быть смещение, возникшее при выборе праймеров; большинство
библиотек, полученных путем РНК-секвенирования падает именно на этом модуле. Обычно это не исправляется какой-либо постобработкой;
тем не менее, подобная ошибка не вносит существенной погрешности в последующие измерения
Модуль Sequence Duplication Levels падает, если неуникальные последовательности составляют >50% от общего числа таковых в библиотеке
В нашем случае это могло возникнуть вследствие анализа небольшой РНК-библиотеки. Думаю, в данном случае этот параметр не является серьезной причиной
для того, что наши чтения плохие и не подлежат дальнейшему анализу (особенно с учетом того, что дупликации последовательностей могли возникнуть
в результате технических ошибок пцр, как артефакты)
Таким образом, будем считать наши риды подлежащими дальнейшему анализу
Задания 2-3. Картирование чтений. Анализ выравнивания
| Input | Output | Программа | Что делает |
| chr8.fasta | *indexed | hisat2-build ../chr8.fasta indexed |
Индексируем файл из предыдущего практикума |
| cleaned.40.fastq | samchr8.2.sam | hisat2 -x indexed -U cleaned.40.fastq -S samchr8.2.sam --no-softclip |
Строим выравнивание референса и ридов; при этом убираем флажок --no-spliced-alignment, тк выравниваем риды, полученные путем секвенирования РНК на ДНК-последовательность (в которой исходно есть интроны) |
| samchr8.2.sam | bam.bam | samtools view samchr8.2.sam -b >> bam.bam |
Переводим выравнивание ридов с референсной последовательностью в бинарный формат |
| bam.bam | sortedbam.bam | samtools sort bam.bam sortedbam |
Сортируем полученное выравнивание по координате начала чтения |
| sortedbam.bam | sortedbam.bam.bai | samtools index sortedbam.bam |
Индексируем отсортированный файл с выравниванием |
Строим выравнивание референса и ридов с помощью программы, указанной в таблице выше
На вход было подано 17 491 ридов, из которых 100% были неспаренными;
346 (1.98%) выровнено 0 раз
17 145 (98.02%) выровнено 1 раз
0 (0.00%) выровнено >1 раза
Задание 4. Подсчет чтений
Будем использовать программу htseq-countИнтересующие нас опции программы:
-f тип файла с выравниванием (формат .sam или .bam; .sam по умолчанию)
-s данные из цепь-специфичного анализа -?
-i атрибут gff, который будет использоваться как feature ID
-m модуль для обработки перекрытия ридов -?
Результаты по подсчету:
no_feature 17143
ambiguous 0
too_low_aQual 0
not_aligned 346
alignment_not_unique 0
| Input | Output | Программа | Что делает |
| sortedbam.bam, gencode.v19.chr_patch_hapl_scaff.annotation.gtf | count.txt | htseq-count -f bam -s yes sortedbam.bam /P/y14/term3/block4/SNP/rnaseq_reads/ gencode.v19.chr_patch_hapl_scaff.annotation.gtf >> count.txt |
Подсчитываем риды (по различным критериям) |
Задание 5. Анализ результатов
Предполагаю, что скрипт count.py должен подсчитывать количество чтений. Его выдача:ENSG00000104738.12 1 ENSG00000253729.3 1Так, по одному чтению упали на гены ENSG00000104738 (MCM4; minichromosome maintenance complex component 4) и
ENSG00000253729 (DNA-PKC; protein kinase, DNA-activated, catalytic subunit)