Исследование полиморфизмов у людей
Часть 1. Подготовка чтений
На вход имеем чтения
экзома. С помощью
программы FastQC сделал анализ качества чтении. По результатам
последние позиции ридов часто имеют неудовлетворительное качество,
поэтому их следует очистить. Кроме того, встречаются последовательности
длиной менее 50, их тоже следуюет убрать.
С помощью программы Trimmomatic проводили очистку
ридов. Команда вызова данной программы:
"java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr.fastq
chrout.fastq TRAILING:20 MINLEN:50". Полученный файл также
проанализировал с помощью FastQC. Произведем сравнение качества ридов
до и после очистки.
Изменение общего количества ридов и длины
последовательностей можно посмотреть в таблице 1. Видим, что
общее количество последовательностей уменьшилось на 122, а наименьшая
длина последовательности увеличилась с 30 до 50.
| Таблица 1. Оценка качества чтений до и после очистки | ||
|---|---|---|
| До | После | |
| Общее количество ридов | 5068 | 4946 |
| Длины последовательностей | 30-100 | 50-100 |
Сравним качество чтений по картинкам "Per base
quality" до и после очистки. Данные картинки приведены ниже. Видим,
что качество после очистки заметно возросло. И лишь по последней
позиции встречается более менее заметный хвост, уходящий в желтую зону.

Картинка 1a. Качество по основаниям до очистки
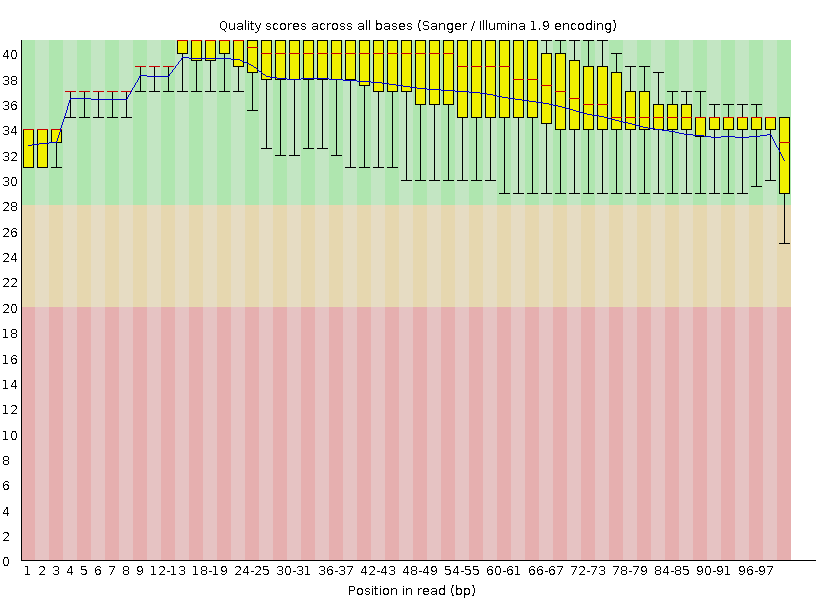
Картинка 1b. Качество по основаниям после очистки
Рассмотрим также другие параметры. Например,
среднее качество чтений по последовательностям и чрезмернопредставленные последовательности.
По первому параметру мы видим лишь незаметное улучшение результатов.
Посмотрим это на картинках ниже. Кривая теперь начинается не с 2,
а с 19 и идет ближе к 0 в начале. Но и там и там наблюдается максимум
в районе 38. Этот пункт показывает нам как много последовательностей
не имеют хорошего среднего качества чтения. Это выражается тяжестью
левого хвоста. Чем тяжелее, тем хуже. Тяжесть хвоста больше до чистки.
Однако в файле после чистки появились чрезмернопредставленные
последовательности. Скорее всего они были и раньше, но не превышали
определенный порог. После того как общее количество ридов было уменьшено
доля этих последовательностей увеличилась и превысила 0.1%. Данные по
этим последовательностям представлены в таблице 2. В хороших чтениях
чрезмернопредставленных последовательностей быть не должно, однако, их
появление еще не говорит о плохом качестве чтения. Вероятность появления
определенной последовательности длины 50 и больше очень мала. Поэтому,
когда мы встречаем такие последовательности в разных частях генома, мы
можем заподозрить что-то неладное. Однако, этот показатель сильно
зависит от общего количества последовательностей. При малом количестве
последовательностей есть вероятность случайно встретить такие
повторяющиеся кусочки. Но при увеличении этого количества вероятность
случайной встречи стремится к нулю.
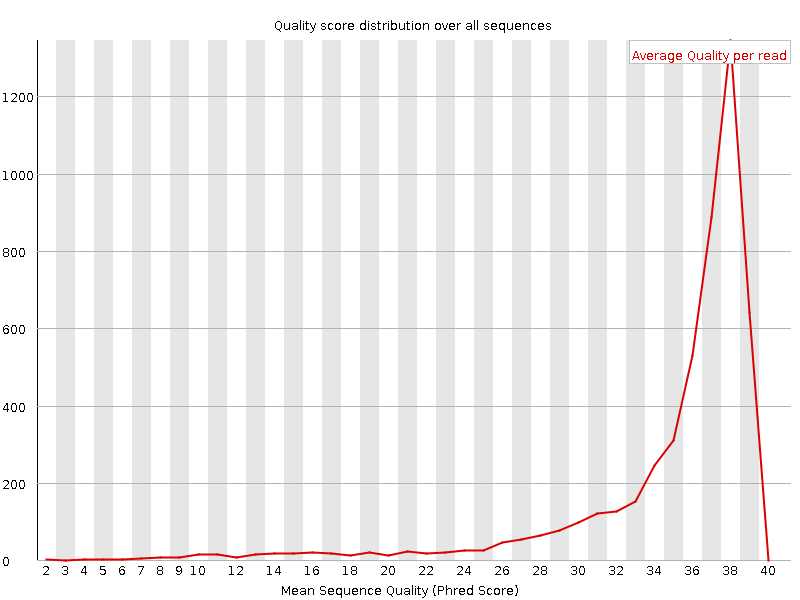
Картинка 2a. Качество по последовательностям до очистки
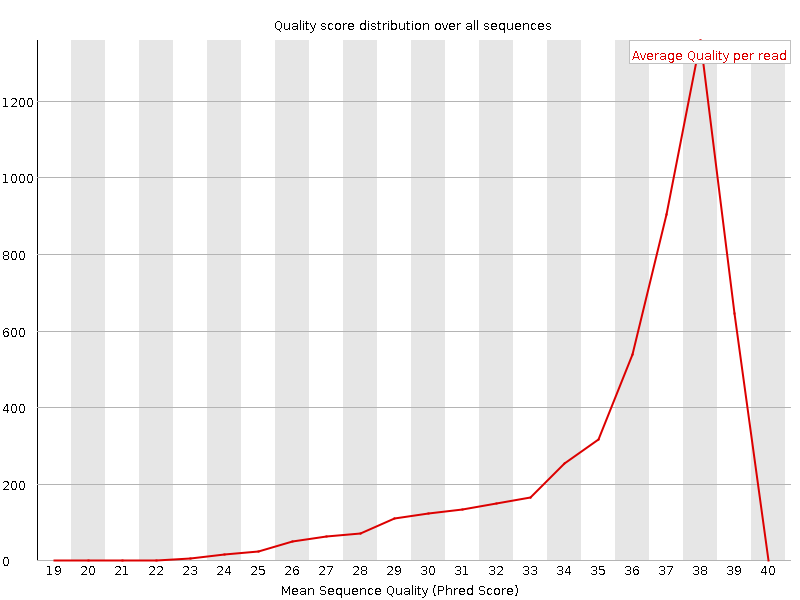
Картинка 2b. Качество по последовательностям после очистки
| Таблица 2. Чрезмернопредставленные последовательности после чистки | |||
|---|---|---|---|
| Последовательность | Количество | Процентный состав | Возможный источник |
TGCGTCTGGAATAAAGTAGTTGTAGCTGGCCTGACTCTTGAAAATTACAG | 5 | 0.10109179134654267 | No Hit |
TAAGAGAACATTACATTTAAAAAGAAAGCAATAACAAAACCCAAGTATAC | 5 | 0.10109179134654267 | No Hit |
CCCACCCTTGCCTGTTCTGTGTGCTACTGCTAACCTCCTGTGGGCTGAGA | 5 | 0.10109179134654267 | No Hit |
AAGATAATGCTTGTAAGTGAATACTGGATAGTGGAGAAAAAACAGAAACA | 5 | 0.10109179134654267 | No Hit |
AATGAGACAAATGTCAAACAAGCACATCAACTTCTTAAAAAACAAAGAAA | 5 | 0.10109179134654267 | No Hit |
AAAAGCAACATTATTATAAATACATATATATGATATGTAGACAGATGGCT | 5 | 0.10109179134654267 | No Hit |
Часть 2.
Картирование чтений.
С помощью команды: "bwa index chr15.fasta" проиндексировал 15 хромосому. Далее командой : "bwa mem chr15. chrout.fastq > read.sam"
Анализ выравнивания.
Анализ проводил с помощью следующих команд: 1)samtools view read.sam -b -o read.bam, -b меняет формат на bam, а -o позволяет выбирать имя выходного файла;
2)samtools sort read.bam -T waste.txt -o sread.bam, -T позволяет записывать промежуточные значение во временный файл, а не в стандартный вывод. Команда сортирует bam файл.
3)samtools index sread.bam. Команда индексирует отсортированный файл.
4)samtools idxstats sread.bam. Команда находит количество откартированных чтений. Результат работы программы. Первая строчка показывает, что 4946 чтений попали на 15 хромосому, а 1 - никуда.
Анализ проводил с помощью следующих команд: 1)samtools view read.sam -b -o read.bam, -b меняет формат на bam, а -o позволяет выбирать имя выходного файла;
2)samtools sort read.bam -T waste.txt -o sread.bam, -T позволяет записывать промежуточные значение во временный файл, а не в стандартный вывод. Команда сортирует bam файл.
3)samtools index sread.bam. Команда индексирует отсортированный файл.
4)samtools idxstats sread.bam. Команда находит количество откартированных чтений. Результат работы программы. Первая строчка показывает, что 4946 чтений попали на 15 хромосому, а 1 - никуда.
Определим среднее покрытие чтениями какого-нибудь экзона. Для начала найдем хорошо покрытые чтениями нуклеотиды.
Для создания гистограммы и вывода таких нуклеотидов написал скрипт. В Genome Browser NCBI
нашел 15 хромосому сборки hg19
Нашел экзон, в которых входит нуклеотид в 77777829 позиции. Границы этого экзона - 77775312..77777945. В результате работы
скрипта
было получено среднее покрытие экзона, оно составило 29.4.
Часть 3. Анализ SNP
Используем следующие команды:
1)samtools mpileup -uf chr15.fasta sread.bam -o snp.bcf. Команда создает файл с полиморфизмами в формате bcf.
2)bcftools call -cv snp.bcf -o snp.vcf. Команда создает файл со списком отличий между референсом и чтениями в формате vcf.
1)samtools mpileup -uf chr15.fasta sread.bam -o snp.bcf. Команда создает файл с полиморфизмами в формате bcf.
2)bcftools call -cv snp.bcf -o snp.vcf. Команда создает файл со списком отличий между референсом и чтениями в формате vcf.
Описание трех полиморфизмов в таблице 3. Общее количество инделей - 3, вставок - 88.
| Таблица 3. Три полиморфизма из файла vcf | |||||
|---|---|---|---|---|---|
| Тип | Координата | В референсе | В чтении | Глубина | Качество |
| Вставка | 58722590 | gcc | gCcc | 3 | 24.4922 |
| Замена | 58723675 | С | Т | 46 | 225.009 |
| Делеция | 58853212 | GACACAC | GACAC | 92 | 217.468 |
Переходим к заключительному этапу: аннотация SNP.
Использовал команды:
1)perl ../../annovar/convert2annovar.pl -format vcf4 snp.vcf > snp.avinput. Перевод snp из vcf-формата в формат входного файла annovar.
2)perl ../../annovar/annotate_variation.pl -out snp.refgen -build hg19 snp.avinput ../../annovar/humandb/. Команда проводит аннотацию по базе refgene. Из 91 в экзоны попали 12, в интроны - 63, в некодирующие РНК - 10, не попали в гены 2, нетранслируемых регионов - 1. Гомозиготных - 55, гетерозиготных замен - 36. Как и ожидалось, большая часть замен попали в интроны.
3)perl ../../annovar/annotate_variation.pl -out snp.138 -build hg19 -dbtype snp138 snp.avinput ../../annovar/humandb/. Аннотация проводится по базе dbsnp. На выходе получаем файлы с указанием SNP, аннотированных в базе и не аннотированных в базе. Первый файл содержит 76 полиморфизмов, из них 45 гомозиготных, 31 гетерозиготных. Во втором файле - 15 полиморфизмов.
4) perl ../../annovar/annotate_variation.pl -filter -out snp.1000g -build hg19 -dbtype 1000g2014oct_all snp.avinput ../../annovar/humandb/. Аннотация по базе 1000g. Выдача похожа на предыдущую. Как и тогда у нас есть 2 файла: в первом - 75, во втором - 16 полиморфизмов.
5)perl ../../annovar/annotate_variation.pl -regionanno -out snp.gwas -build hg19 -dbtype gwasCatalog snp.avinput ../../annovar/humandb/. Аннотация по каталогу gwas дает интересную выдачу. В этом файле указаны ассоциированые с болезнями полиморфизмы нашего пациента. Файл имеет 5 ассоциаций: 2 из них связаны с липопротеинами высокой плотности холестерина, по одному - с ростом, диабетом 2 типа и последний - с гематологическими и биохимическими чертами.
6)perl ../../annovar/annotate_variation.pl -filter -out snp.clinvar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/. Аннотация по базе данных clinvar. К сожалению, все SNP были отфильтрованы. Пустой первый файл, второй файл, содержащий все SNP.
Использовал команды:
1)perl ../../annovar/convert2annovar.pl -format vcf4 snp.vcf > snp.avinput. Перевод snp из vcf-формата в формат входного файла annovar.
2)perl ../../annovar/annotate_variation.pl -out snp.refgen -build hg19 snp.avinput ../../annovar/humandb/. Команда проводит аннотацию по базе refgene. Из 91 в экзоны попали 12, в интроны - 63, в некодирующие РНК - 10, не попали в гены 2, нетранслируемых регионов - 1. Гомозиготных - 55, гетерозиготных замен - 36. Как и ожидалось, большая часть замен попали в интроны.
3)perl ../../annovar/annotate_variation.pl -out snp.138 -build hg19 -dbtype snp138 snp.avinput ../../annovar/humandb/. Аннотация проводится по базе dbsnp. На выходе получаем файлы с указанием SNP, аннотированных в базе и не аннотированных в базе. Первый файл содержит 76 полиморфизмов, из них 45 гомозиготных, 31 гетерозиготных. Во втором файле - 15 полиморфизмов.
4) perl ../../annovar/annotate_variation.pl -filter -out snp.1000g -build hg19 -dbtype 1000g2014oct_all snp.avinput ../../annovar/humandb/. Аннотация по базе 1000g. Выдача похожа на предыдущую. Как и тогда у нас есть 2 файла: в первом - 75, во втором - 16 полиморфизмов.
5)perl ../../annovar/annotate_variation.pl -regionanno -out snp.gwas -build hg19 -dbtype gwasCatalog snp.avinput ../../annovar/humandb/. Аннотация по каталогу gwas дает интересную выдачу. В этом файле указаны ассоциированые с болезнями полиморфизмы нашего пациента. Файл имеет 5 ассоциаций: 2 из них связаны с липопротеинами высокой плотности холестерина, по одному - с ростом, диабетом 2 типа и последний - с гематологическими и биохимическими чертами.
6)perl ../../annovar/annotate_variation.pl -filter -out snp.clinvar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/. Аннотация по базе данных clinvar. К сожалению, все SNP были отфильтрованы. Пустой первый файл, второй файл, содержащий все SNP.
© Maximov Vladislav, 2019.