| Главная страница | Обучение | Обо мне | Ссылки |
Предсказание генов эукариот | |||
|
Задание 1. Предсказание генов эукариотического организма с помощью ресурса AUGUSTUS Для предсказания генов в эукариотическом геноме X5 был выбран unplaced-717.fasta, чья длина составляет 46913 пн. Предсказание планировалось делать с помощью ресурса AUGUSTUS. Для этого необходимо либо обучить данную программу распознавать гены эукариот (для этого используются гомологи хорошо изученных генов или транскриптомы), либо использовать предсказание генов близкородственного организма как основу для предсказания генов интересующего (неизвестного) организма. Был выбран второй путь. Для этого сначала нужно было найти близкого родственника организма с геномом X5. Для поиска организма, близкого Х5, который подошел бы для обучения AUGUSTUS, для последовательности unplaced-717.fasta был запущен BLAST, алгоритм blastx, который читает входную нуклеотидную посл-ть в 6 рамках считывания и ищет по полученным посл-тям белковых гомологов.
Таким образом, он может найди кодируемые входной посл-тью белки. Параметры blastx: ограничение по таксону (были выбраны только fungi, так как несколько запусков blast для других скэффолдов показали, что большинство находот принадлежит этому таксону), исключение Models (XM/XP) и Uncultured/environmental sample sequences.
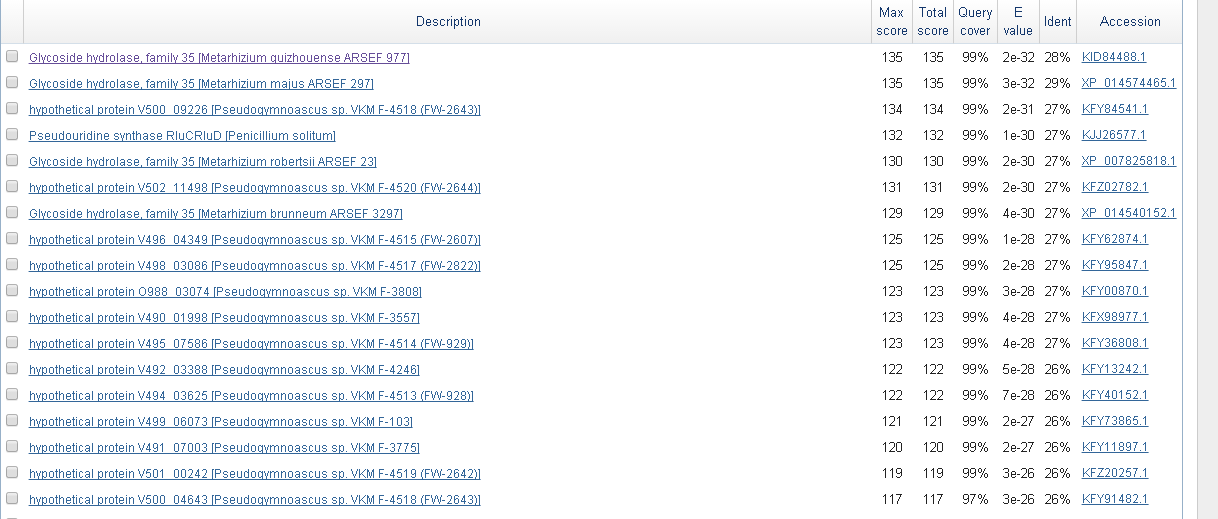
Несколько лучших находок принадлежали представителям рода Rhizopus (рис. 1), а также Spizellomyces, который входит в родственный порядок с Rhizopus. Поэтому для обучения был выбран организм Rhizopus oryzae.
Рис. 1. Фрагмент списка находок blastx для посл-ти unplaced-717.fasta. Затем было проведено предсказание генов в этом контиге с помощью AUGUSTUS. Результаты доступны по ссылке, оттуда же был скачан архив predictions.tar.gz. В нем содержатся несколько интересующих нас файлов. Описание файлов в архиве, полученном в результате предсказания: Проверка предсказания на уровне белков
Для проверки случайным образом были выбраны гены g1, g2, g5, g9 и g4. Предсказание проверялось с помощью алгоритма blastp.
1. g1
Результаты оказались не очень благоприятными: на рис. 2 приведены все находки, полученные blastp. Было проведено 2 поиска - по базе данных nr и по SwissProt. Оба варианта дали не очень высокие параметры выравниваний.
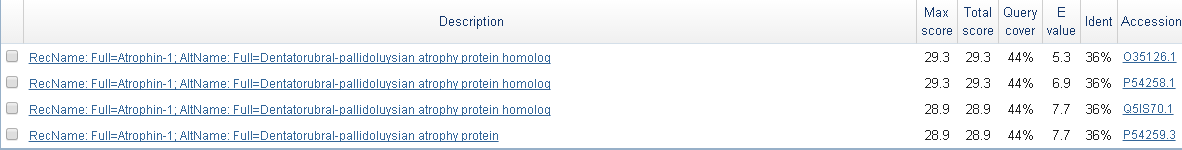
Кроме того, по nr получается, что белок, кодируемый геном g1, гомологичен экзонуклеазе II из организма Schizosaccharomyces octosporus yFS286 (рис. 2а), а по SwissProt (рис. 2b) - Dentatorubral-pallidoluysian atrophy protein из организмов млекопитающих (мышь, крыса, человек и др.). Такие находки назвать достоверными нельзя.
Рис. 2b. Фрагмент списка находок blastp для посл-ти g1.fasta. Наверху показан результат поиска по SwissProt. 2. g2
Ситуация с данным предполагаемым белком обстоит еще хуже: гомологи не были найдены ни по одной базе данных. AUGUSTUS полностью ошибся.
3. g5
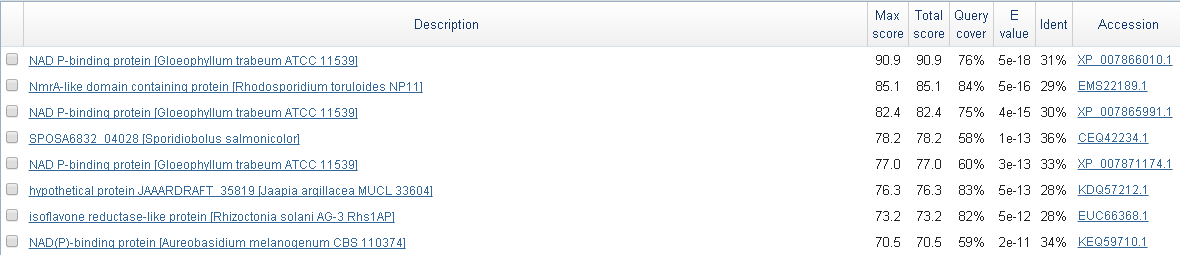
С этим белком повезло больше: нашлось много гомологов с неплохими параметрами query cover и e-value, самые лучшие из них (приведена на рис. 3) принадлежат Fungi и имеют похожие названия.
Учитывая полученные результаты построения доменной структуры и находки бласта, можно заключить, что белок, закодированный в 5-ом гене, является НАДФ-связывающим.
4. g9
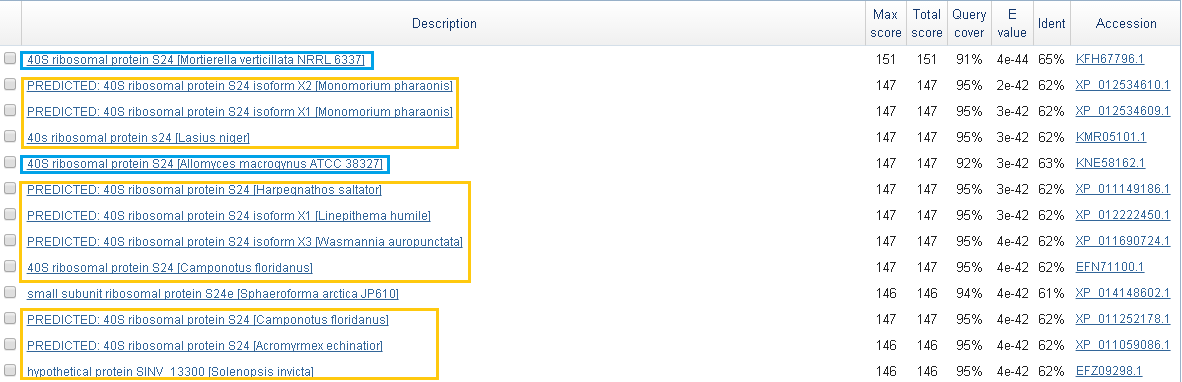
Он аннотировался хорошо, много достоверных находок, но ... они принадлежат в основном муравьям. См. рис.4. Возможно, дело в том, что это 40S-рибосомальный белок. Рибосомальные белки сами по себе являются очень консервативными структурами, поэтому можно предположить вероятность, что даже у настолько филогенетически дальних родственников, как муравей и гриб, эти белки будут сильно схожи.
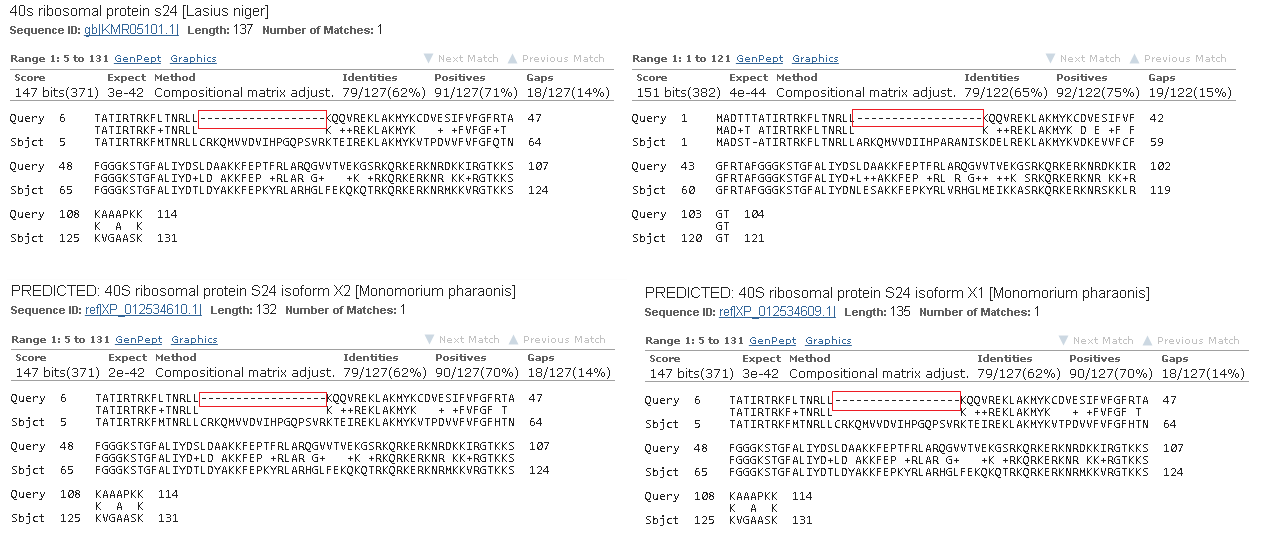
Для всех выравниваний характерно наличие одного гэпового участка длиной 18 позиций в query. Отсутствующий фрагмент может указывать на то, что AUGUSTUS не нашел один экзон, который должен был быть в мРНК. "Гэповую архитектуру" выравнивания можно увидеть на рис. 5.
5. g4
Судя по результатам blastp (рис. 6), ситуация довольно интересная: высокий query cover, но очень низкое сходство (в общем по находкам Identity < 50%). Тем не мнеее, белок определен как гликозилгидролаза, если верить самым первым находкам, а если не верить им, то как он как минимум входит в семейство белков с альфа-амилазным доменом.
Это подтверждается построенной доменной структурой (рис. 6b), а также описанием гипотетического белка из организма Pseudogymnoascus sp. разных штаммов. Оно приведено на рис. 7. Видно, что в нем есть уже упоминавшийся домен.
Так как покрытие последовательности почти 100% в большинстве случаев, а score низкий, можно заподозрить то, что для этого белка была неверно определена экзон-интронная структура. Это заметно по большим фрагментам гэпов в "хороших" выравниваниях (см. рис. 8).
Такой вид выравнивания присущ не только первой находке в списке Blast, но и всем похожим находкам. Скорее всего, при аннотации четвертого гена была нарушена экзон-интронная структура. Это подтверждается тем, что "рисунок" гэпов в выравниваниях примерно одинаков (для примера приведено еще одно выравнивание на рис. 8).
Задание 2. Сравнение аннотации RefSeq и AUGUSTUS для одного гена человека
В качестве проверяемого гена человека был взят ген OGDH, кодирующий фермент 2-оксоглутаратдегидрогеназу. Его Gene ID в NCBI - 4967. Ген закодирован на +-цепи 7 хромосомы, координаты chr7:44606522-44709070 (и размер гена 102549 пн). Использовался ресурс USCS Genome Browser.
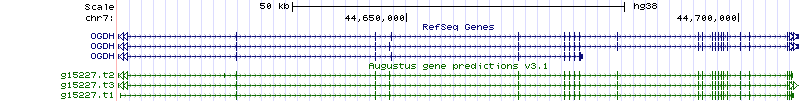
На рис. 9 показано окно Genome Browser с изображением экзон-интронной структуры гена OGDH с тремя трэками: base position, RefSeq и AUGUSTUS.
Рис. 9. Изображение экзон-интронной структуры гена OGDH. При обработке аннотаций RefSeq и AUGUSTUS в табличной форме я столкнулась с проблемой: в RefSeq по Genome Browser было дано целых 3 разных аннотации с идентификаторами NM_002541, NM_001165036, NM_001003941, для них было указано 23, 23 и 9 экзонов соответственно. По-видимому, это были аннотации для разных изоформ белка OGDH. Я проверила каждый из идентификаторов в NCBI, и там было сказано, что на данный момент в этом гене насчитывают 26 экзонов. Таким образом, изоформы, содержащей в себе все количество экзонов, hg38 не представлено.
Для дальнейшей работы я оставила аннотацию с идентификатором NM_002541 и 23 экзонами. Аннотация AUGUSTUS была проведена по 3 транскиптам. Количество полученных экзонов различается: в первом (AUGUSTUS t1) аннотировано 23 экзона, во втором (AUGUSTUS t2) - 24, а в третьем (AUGUSTUS t3) аж 41 экзон.
Итак, были получены таблицы с аннотацией RefSeq и AUGUSTUS. Они были сведены в один файл: Таблица с аннотациями RefSeq и AUGUSTUS.
некоторые выводы о различиях двух приведенных аннотаций:
В целом хочется отметить, что аннотации с помощью RefSeq и AUGUSTUS не внесли ясности в экзон-интронную структуру данного гена. Не очень также понятно, какой из этих БД можно доверять больше.
| |||
| © Alexandra Boyko, 2014. Faculty of Bioengineering and Bioinformatics, MSU. | |||