| Главная страница | Обучение | Обо мне | Ссылки |
EMBOSS: Пакет программ для анализа последовательностей | |||
|
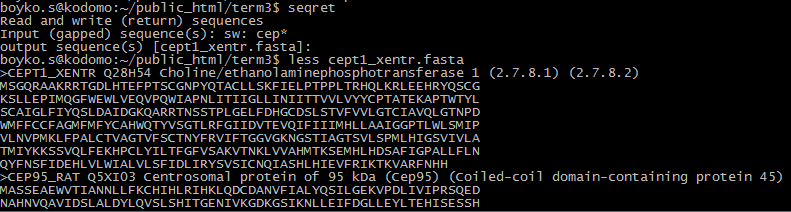
В данном практикуме было необходимо освоить некоторые программы из пакета EMBOSS. Упражнения 1. seqret. Программа читает файлы и записывает их в общий файл (по умолчанию формата fasta). На скриншоте видно, что на вход (input sequence(s)) были поданы последовательности из банка SwissProt, чей идентификатор начинается с CEP. Все последовательности были объединены в файл cept1_xentr.fasta, его начальный фрагмент также виден на скриншоте. (Общий файл оказывается назван по идентификатору первой последовательности.)
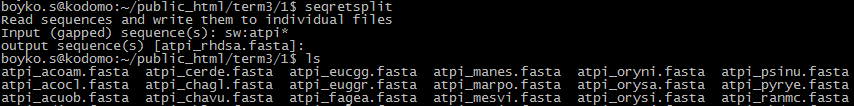
2. seqretsplit. Программа читает файлы и записывает их в отдельные fasta-файлы. На скриншоте показано, что в качесвте входных последовательностей были поданы записи SwissProt'а, чьи идентификаторы начинаются с ATPI. Программа создала файлы, каждый из которых назван по идентификатору посл-ти, лежащей в нем. Часть списка созданных файлов представлена на скриншоте.
3. transeq. Программа транслирует нуклеотидные посл-ти из общего fasta-файла в аминокислотные посл-ти, которые тоже сохраняются в одной файле. На вход был подан файл all_nucl.fasta с 5 нуклеотидными последовательностями (гистоны H3 находок из прошлого практикума). После трансляции аминокислотные посл-ти были отправлены в файл all_pep.fasta. Его фрагмент представлен на втором скриншоте.
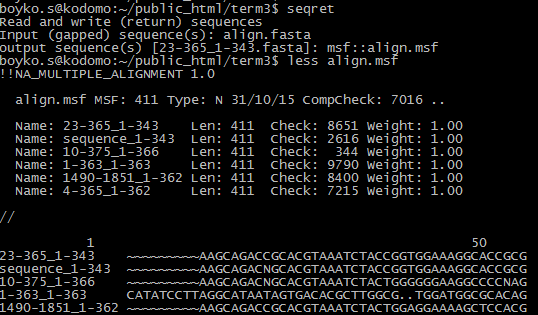
4. seqret. Эта програма также может менять формат интересующего нас файла. В данном примере (см. скриншот) было взято выравнивание нескольких нуклеотидных последовательностей в формате (align.fasta), затем оно было переведено в формат msf. Фрагмент полученного файла (align.msf) также приведен на скриншоте.
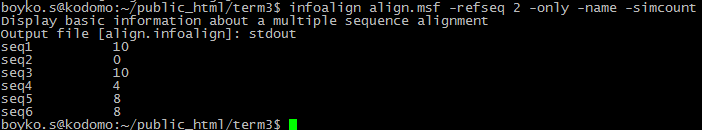
5. infoalign. Программа выдает самую разнообразную информацию о выравнивании. В данном случае, было взято выравнивание align.msf (с откорректированными для удобства названиями посл-тей). Интересовало количество совпадающих букв в посл-ти seq2 и всех остальных. Результат был выведен в stdout (см. скриншот). Были введены дополнительные условия -only -name -simcount, чтобы результат был показан в виде "имя последовательности, кол-во совпадающих букв".
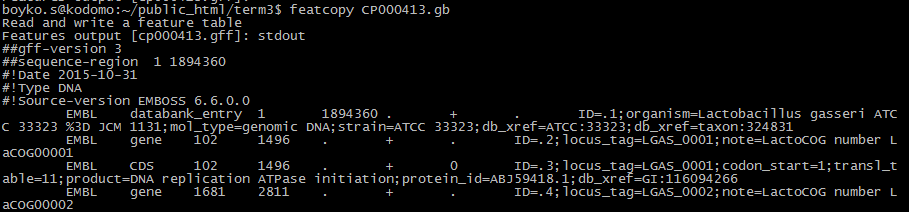
6. featcopy. Данная программа считывает и выдает таблицу особенностей из записи какой-либо последовательности в том формате, который требует пользователь. На скриншоте приведен пример использования featcopy для получения таблицы особенностей генома бактерии Lactobacillus gasseri (CP000413) в формате gff. Сначала запись была скачана в формате gb (genbank) с сайта NCBI. На скриншоте ниже приведено начало полученной таблицы в формате gff.
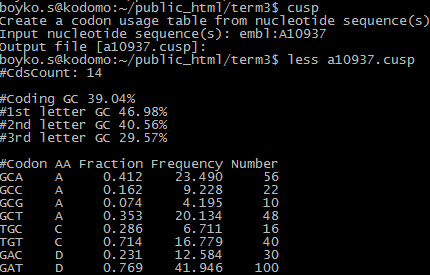
7. cusp. Эта программа рассчитывает частотности использования кодонов в нуклеотидной последовательности и выдает результат в виде файла с таблицей. На вход была подана запись A10937 генома человеческого риновируса 89 (HRV89). Результат частично приведен на скриншоте. Интересно, что помимо собственно частоты использования того или иного кодона, программа выдает процент содержания GC, абсолютное количество появления в посл-ти всех кодонов и их соответствие аминокислотам.
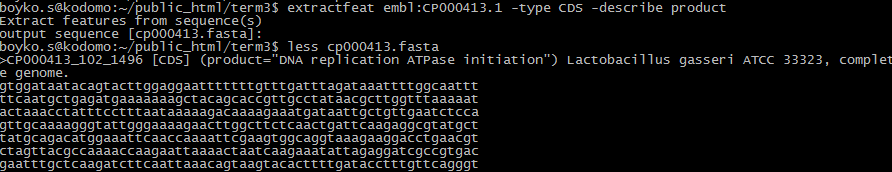
8. extractfeat. Программа читает файл с последовательностями и выдает из них интересующие фрагменты. Например, из генома бактерии Lactobacillus gasseri CP000413 можно получить кодирующие последовательности: запрос программы с параметрами приведен на скриншоте ниже. Часто может быть нужен параметр -join, позволяющий объединить несколько экзонов, относящихся к одному гену, в общую кодирующую последовательность. В случае бактерии он не нужен.
С помощью параметра -describe product можно добавить в получаемый файл описание продукта каждой кодирующей последовательности. Фрагмент выдачи также представлен на скриншоте. 9. tranalign. Программа выравнивает нуклеотидные последовательности исходя из выравниваний их продуктов.
В файле gen_align.fasta содержится выравнивание двух генов из файла gen_seq.fasta, построенное с помощью выравнивания белков-продуктов выбранных генов prot_align.fasta. Сравнение аннотации генов белков археи Pyrobaculum oguniense TE7 с трансляциями длинных открытых рамок считывания Для выполнения этого задания была выбрана архея, которую я изучала в первом семестре. Некоторая информация о ней представлена на странице Pyrobaculum oguniunse TE7. Для археи Pyrobaculum oguniense в GenBank представлена запись по единственной хромосоме: AC - CP003316. Последовательность этой хромосомы была сохранена с аннотациями в формате gb: CP003316.gb Сначала были получены трансляции открытых рамок считывания: с помощью программы getorf с параметрами, приведенными на скриншоте. Файл с трансляциями: cp003316.fasta. Затем был получен список координат и ориентаций найденных открытых рамок: сначала была использована программа infoseq с параметрами, приведенными на скриншоте ниже.
После этого полученная таблица была отредактирована в Excel. Ее итоговый вид: cp003316_orf.xls. Это список трансляций длинных открытых рамок считывания этой хромосомы. Далее я получила таблицу аннотированных белков этой хромосомы, скачав ptt-файл и немного трансформировав его. Итоговая таблица: cp003326_annot.xls. Так же были скачаны последовательности всех белков хромосомы Pyrobaculum oguniense. Они хранятся в файле Pyr_proteins.faa. Таблицы трансляций cp003316_orf.xls и аннотированных белков cp003326_annot.xls были объединены в общую Таблицу. Анализ таблицы Полученная таблица состоит из 19512 строк (т.е. потенциальных или реальных белков), из них всего 2800 белков аннотированы, то есть почти 1/7-ая, что означает, что реально существующих белков у этой археи в 6 раз меньше, чем существует orf'ов. С помощью сортировки таблицы по длине было выяснено, что существуют (аннотированы) белки Pyrobaculum oguniense, которые короче 60 амк (то есть их ген короче 180 пн). Таких белков целых 146 штук, и мы не могли найти их программой getorf с такими параметрами, как мы задали. Примеры приведены на рис. 1.
Расхождения в открытых рамках считывания и координатах генов аннотированных белков требуют отдельного обсуждения. Чаще всего, эти расхождения возникают из-за того, что открытые рамки считавания находятся программой getorf от стоп- до стоп-кодона, причем не включая последний стоп-кодон. Естественно, старт-кодон гена белка находится между двумя стоп-кодонами, а сам ген включает в себя свой стоп-кодон. Поэтому координата конца орфа на 3 меньше, чем координата стоп-кодона гена белка. Примеры приведены на рис. 2.
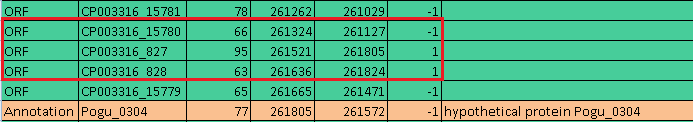
Еще весьма частой проблемой является отсутствие соответствующего гена белка для какой-либо открытой рамки считывания. Примеры приведены на рис. 3.
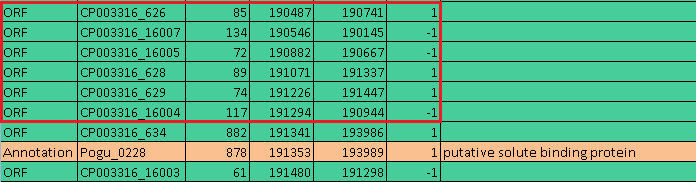
Очень часто может возникнуть ситуация, когда программа getorf с параметром поиска "от стоп- до стоп-кодона" не найдет самую первую кодирующую последовательность, так как она скорее всего начинается до первого стоп-кодона, который встретится в ДНК. В моем случае на прямой цепи этой проблемы не возникло, а на комплементарной цепи хоть и есть ген, начинающийся с первого нуклеотида (вернее, им заканчивающийся, так как он идет в обратном направлении) - Pogu_0001, однако для него нашлась рамка считывания - см. Рис. 4.
В данном геноме можно найти перекрытие рамок длиной более 150 пн. Например, на рис. 5 представлены такие две рамки с перекрытием рамок считывания на прямой и обратной цепях длиной в 230 пар нуклеотидов. Однако ни та, ни другая рамка не содержат гена, про них нельзя сказать ничего особенного.
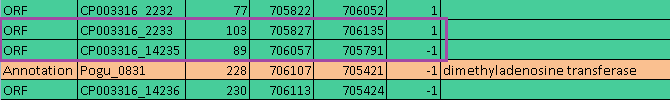
Интереснее ситуация выглядит в другом случае: на рис. 6 представлены две перекрывающиеся на 373 нуклеотида рамки считывания (верхняя строчка и нижняя строчка), а также ген, имеющийся в одной из них. Ген присутствует в более длинной рамке.
| |||
| © Alexandra Boyko, 2014. Faculty of Bioengineering and Bioinformatics, MSU. | |||