Докинг низкомолекулярных лигандов в структуру белка
В этом практикуме мы будем проводить докинг разных лигандов в структуру лизоцима жабы, которая была определа в прошлом практикуме. Делать это будем с помощью пакетов Autodock Vina и oddt. Для работы возьмем третью модель BUFGA.pdb (у нее была скор 1074).
import numpy as np
import copy
# Отображение структур
import IPython.display
import ipywidgets
from IPython.display import display,display_svg,SVG,Image
# Open Drug Discovery Toolkit
import oddt
import oddt.docking
import oddt.interactions
# Органика
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
import pmx # Модуль для манипулирования pdb
pdb = pmx.Model('BUFGA.pdb')
Отделим лиганд от аминокислотных остатков белка: найдем в пдб-файле остатки лиганда и разделим информацию на 2 пдб-файла: только про белок и только про лиганд.
for r in pdb.residues[135:]:
print r #посмотрим остатки
prot = pdb.copy()
for r in prot.residues[-3:]:
prot.remove_residue(r)
lig = pdb.copy()
for r in lig.residues[:-3]:
lig.remove_residue(r)
prot.writePDB('BUFGA_prot.pdb')
lig.writePDB('lig.pdb')
Находим геометрический центр лиганда:
coords = np.zeros((len(lig.atoms),3))
for c,a in enumerate(lig.atoms):
coords[c,:] = a.x
geom_center = np.mean(coords, axis = 0)
geom_center
Подготовка белка для докинга:
prot = oddt.toolkit.readfile('pdb','BUFGA_prot.pdb').next()
prot.OBMol.AddPolarHydrogens()
prot.OBMol.AutomaticPartialCharge()
print 'is it the first mol in BUFGA_prot is protein?',prot.protein,':) and MW of this mol is:', prot.molwt
Странно, что oddt считает, что в BUFGA_prot.pdb не белок, потому что ничего другого там нет.
! less BUFGA_prot.pdb
Затем создаем набор лигандов для докинга на основе известного NAG.
smiles = ['O=C(NC1C(O)C(O)C(OC1O)CO)O', 'O=C(NC1C(O)C(O)C(OC1O)CO)[NH3+]',
'O=C(NC1C(O)C(O)C(OC1O)CO)', 'O=C(NC1C(O)C(O)C(OC1O)CO)(c1ccccc1)',
'NC1C(C(C(OC1O)CO)O)O', 'NC1C(C(C(OC1O)CO)O)OC',
'O=C(NC1C(O)C(O)C(OC1O)CO)C(=O)O', 'O=C(NC1C(C)C(C)C(OC1O)CC)C', 'O=C(NC1C(O)C(O)C(OC1O)CO)C']
mols= []
images =[]
for s in smiles:
m = oddt.toolkit.readstring('smi', s)
if not m.OBMol.Has3D():
m.make3D(forcefield='mmff94', steps=150)
m.removeh()
m.OBMol.AddPolarHydrogens()
mols.append(m)
###with print m.OBMol.Has3D() was found that:
### deep copy needed to keep 3D , write svg make mols flat
images.append((SVG(copy.deepcopy(m).write('svg'))))
display_svg(*images)
Сделала NAG (последняя структура), несколько его модификаций с заменой метильного радикала СH3C(=O)NH группы, а также лиганд с тремя метильными радикалами вместо ОН в сахаре (предпоследняя структура).
Дальше проводим докинг всех этих лигандов в жабий лизоцим. Центром докинга сделаем найденный геометрический центр лиганда.
# create docking object
dock_object = oddt.docking.AutodockVina.autodock_vina(
protein = prot,size=(20,20,20), center = geom_center,
executable = '/usr/bin/vina', autocleanup = True, num_modes = 20)
print dock_object.tmp_dir
print " ".join(dock_object.params)
# do it
res = dock_object.dock(mols, prot)
Визуализация результатов докинга
import pandas as pd
# отсортируем результаты по аффинности (параметр vina_affinity):
res_sorted = sorted(res, key = lambda x : float(x.data['vina_affinity']))
for i,r in enumerate(res):
print "%4d%10s%8s%8s" %(i, r.formula, r.data['vina_affinity'], r.data['vina_rmsd_ub'])
hbtotal = []
hbstrict = []
phob = []
for i,r in enumerate(res_sorted):
int1, int2, strict = oddt.interactions.hbonds(prot,r)
hbtotal.append(len(int1))
hbstrict.append(strict.sum())
ph1, ph2 = oddt.interactions.hydrophobic_contacts(prot,r)
phob.append(len(ph1))
sort_table = pd.DataFrame({'Formula':[r.formula for r in res_sorted],
'Affinity, kcal/mol':[r.data['vina_affinity'] for r in res_sorted],
'Total number of h-bonds':hbtotal, 'Strict number of h-bonds':hbstrict, 'Hydrophobic':phob,
'RMSD':[r.data['vina_rmsd_ub'] for r in res_sorted]})
sort_table[:]
Самым высокоаффинным лигандом оказался C13H17NO6 (r0.pdb) - это лиганд с фенильной группой вместо метильного радикала. Посмотрим в PyMol его, а также NAG (C8H15NO6, r7.pdb) и еще парочку из представленных выше (C11H21NO3 r55.pdb и C7H15NO5 r80.pdb, который является наименее аффинным, если судить по таблице).
for i in (0, 7, 55, 80):
res_sorted[i].write(filename='res%s.pdb' % str(i), format='pdb')
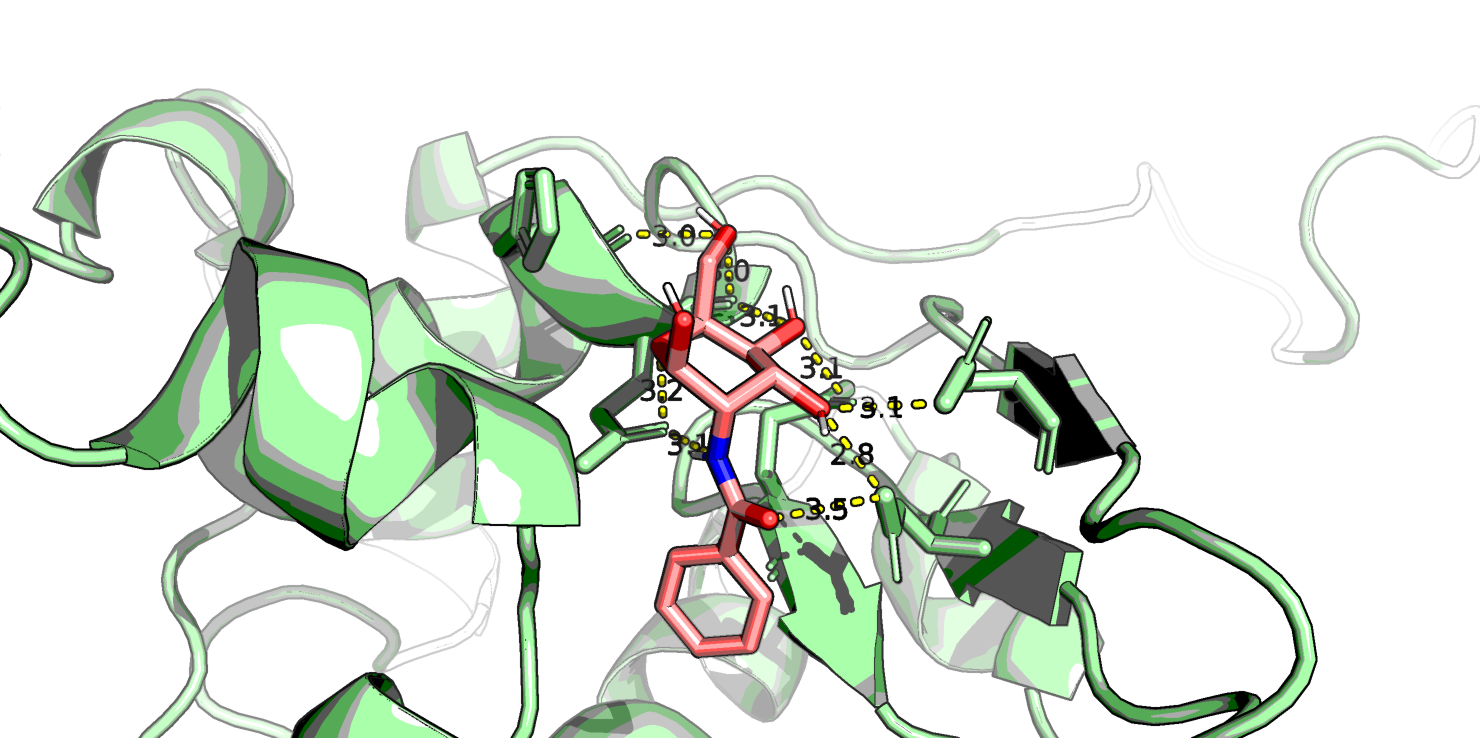

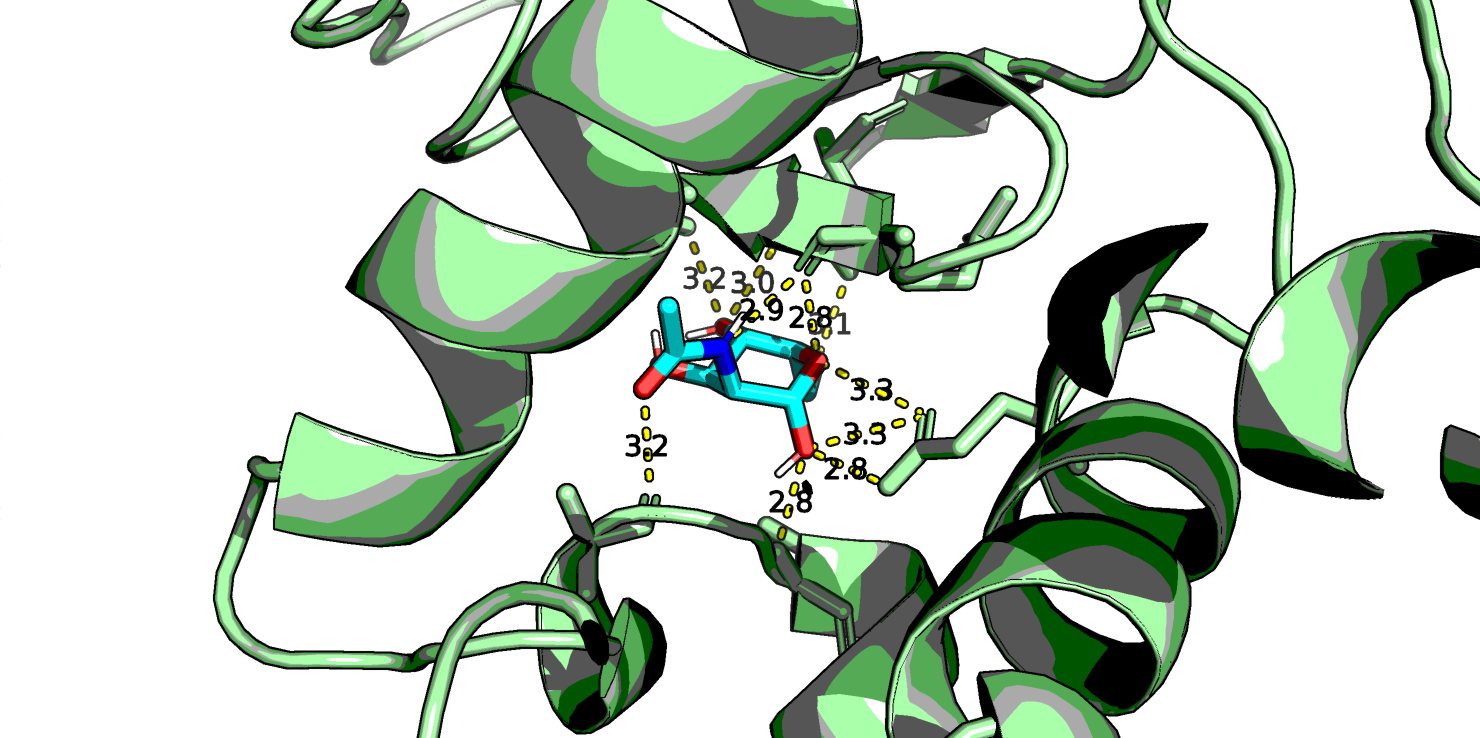
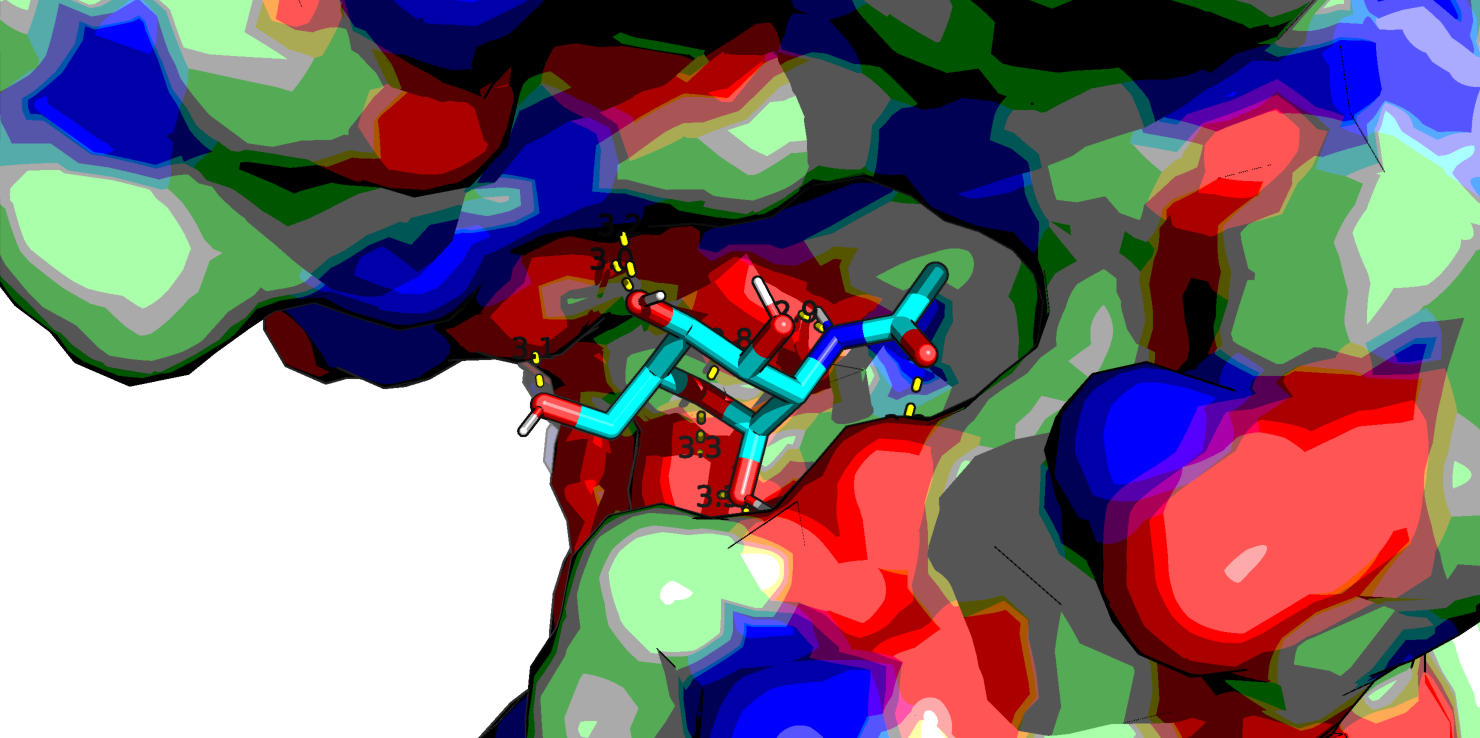
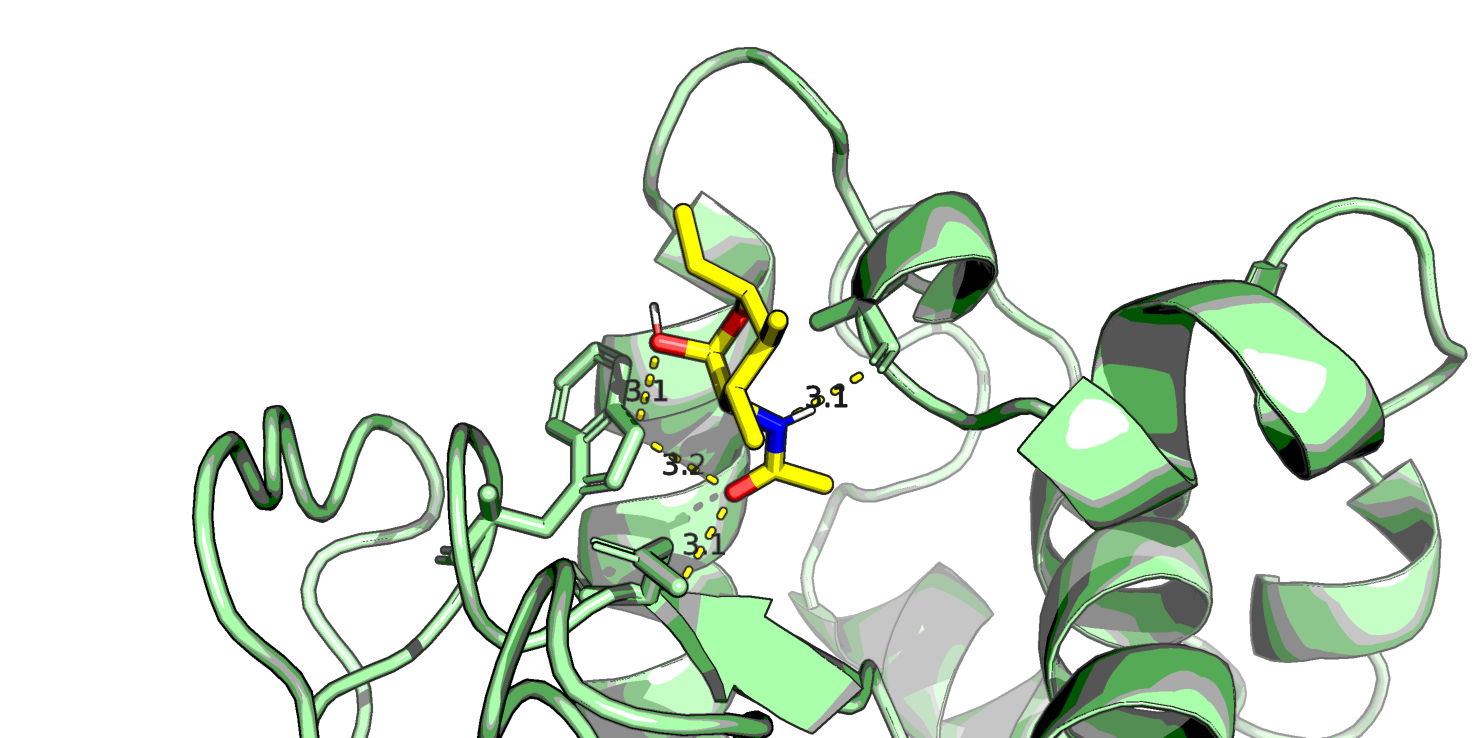
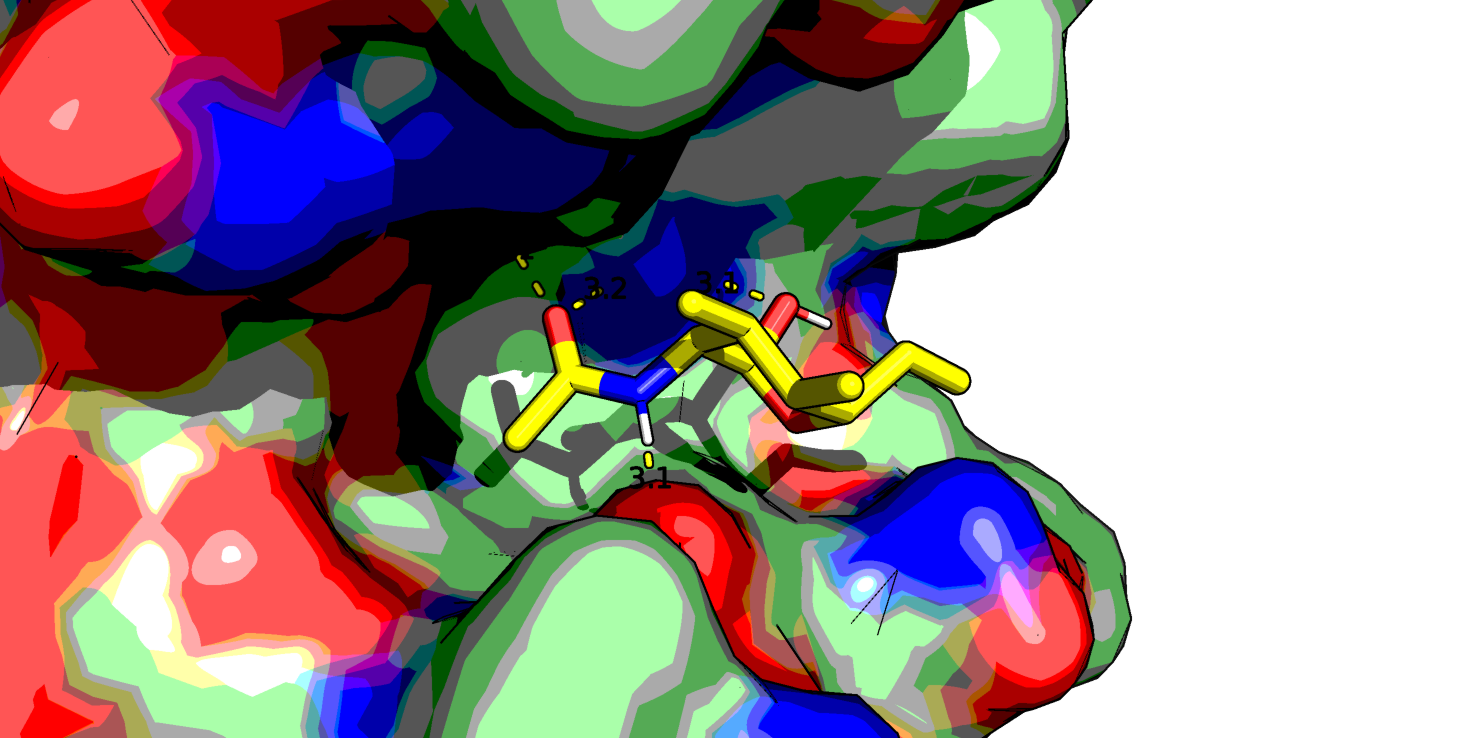
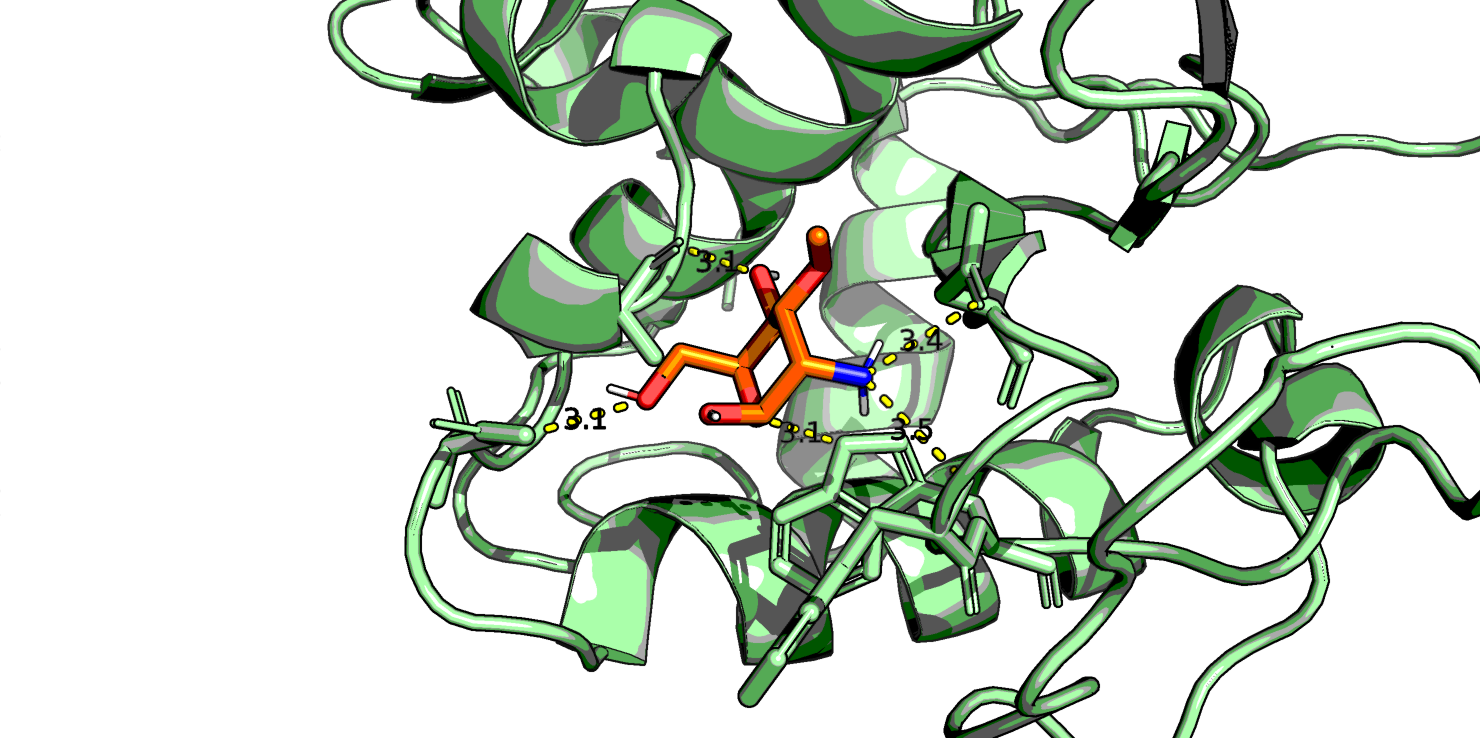
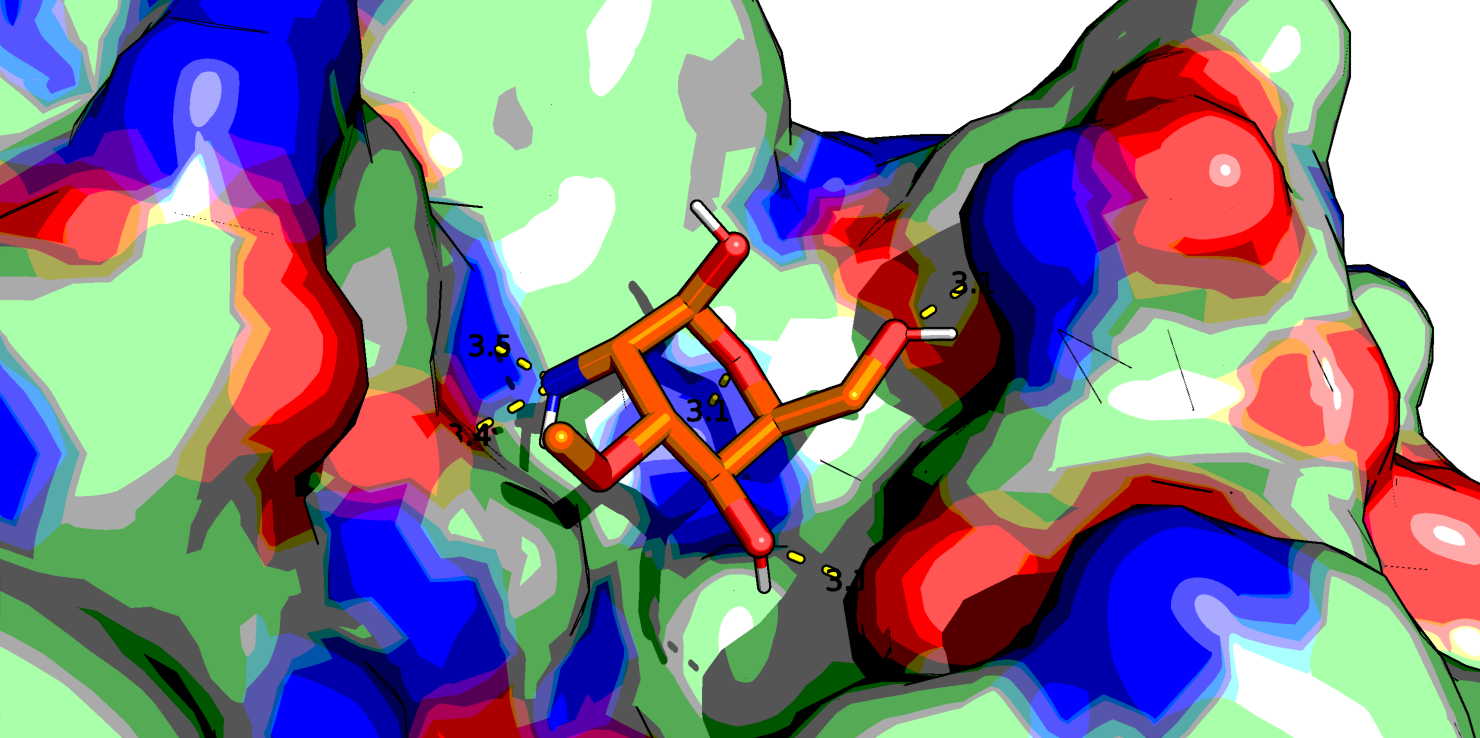
Глюкоза у всех четырех лигандов находится в конформации "кресло". Первый лиганд хорошо ложится в "карман" белка и, вероятно, стабилизируется стэкинговым взаимодействием бензольного кольца с аминокислотными остатками белка, поэтому у него высокая аффинность. Видно, что нативный лиганд NAG взаимодействует с белком большим количеством водородных связей, правда, в одном месте кислород ОН-группы образует аж 3 водородные связи - что-то многовато даже для кислорода. У последнего лиганда азот NH2-группы тоже почему-то образует 2 водородные связи с белком.