Нуклеотидный blast
Задание 1. См. Задание 4 практикума №7.
Задание 2. 3 алгоритма blast.
В этом задании требовалось для последовательности из практикума 7 (последовательность) запустить 3 разных алгоритма blast (blastn, megablast, discontiguous megablast) и сравнить результаты их работы.
Для каждого запуска были установлены следующие параметры:
- Было решено ограничить находки тем же родом, что и лучшая находка в Задании 1, а именно Polycirrus (область поиска не удалось ограничить конкретным видом, получалось слишком мало находок; а при выборе другого рода процент сходства был слишком низкий).
- Количество выводимых находок я ограничила 1000 (по умолчанию было 100), чтобы отследить их реальное количество.
- Все остальные параметры по умолчанию и одинаковы для всех 3 запусков.
В Таблице 1 представлены некоторые характеристики результатов работы 3 алгоритмов.
Таблица 1. Сравнение 3 алгоритмов BLAST.
| Название алгоритма | blastn | megablast | discontiguous megablast |
| Число находок | 34 | 4 | 27 |
| E-value худшей находки | 2.5 (ген рРНК - короткая последовательность)
2e-147 (нужный ген - большой процент покрытия) |
0.0 | 2e-147 |
| Процент сходства для худшей находки | 100% (короткая)
77% (полная) |
77% | 89% |
| Скриншот страницы находок | Рис. 1а | Рис. 1b | Рис. 1с |
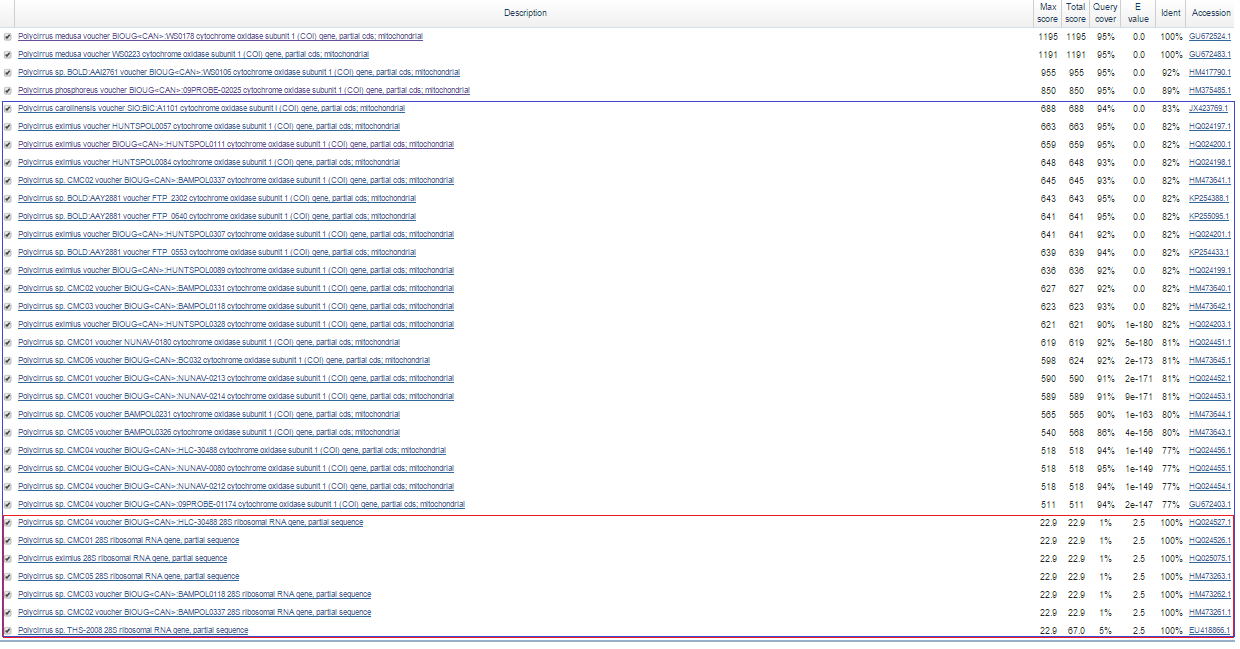
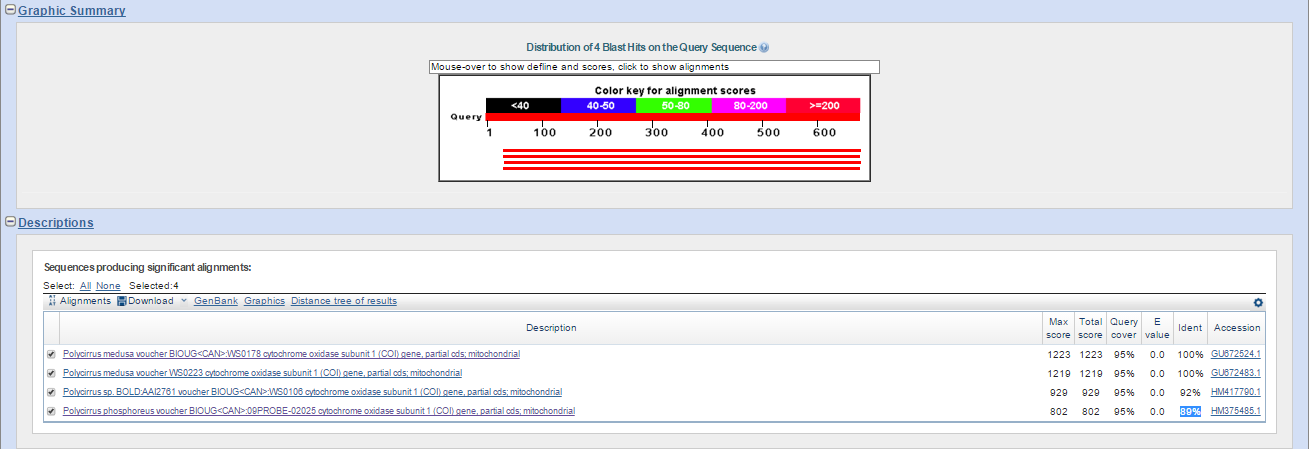
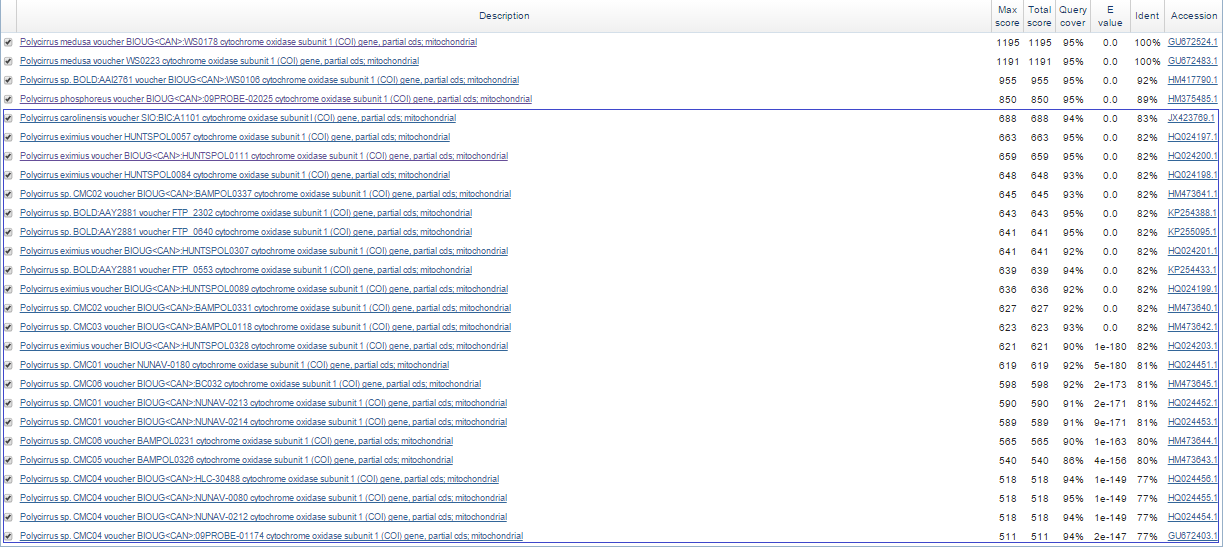
Как видно из данных Таблицы 1 и Рис.1а-с, набор находок неодинаков для разных алгоритмов.
- В частности, megablast нашел только 4 наиболее достоверные находки с наименьшим E-value (0.0) и наибольшим процентом сходства (100%-92%). Этот алгоритм позволяет найти последовательности, обладающие очень высоким сходством с исходной.
- discontiguous megablast нашел на 23 находки больше (Рис. 2а), например, 4 находки из организма Polycirrus eximius (E-value = 0.0, процент сходства - 82%). Как позднее будет видно в сравнении с выдачей blastn, discontiguous megablast выдал все находки, которые можно считать достоверными: они относятся к тому же гену, который представлен в исходной последовательности. megablast не нашел эти последовательности, так как он ищет сходные последовательности по паттерну в 28 нуклеотидов (выше вероятность попадания замены, чем в паттерне длиной 11 нуклеотидов).
- blastn нашел еще 7 находок (в сравнении с discontiguous megablast), которые относятся к участкам последовательностей генов рРНК и являются очень короткими (11-12 нуклеотидов): эти находки имеют худший E-value = 2.5 и покрытие только 1% (Рис. 2b). Из этого следует, что blastn ищет любые сходные участки, а discontiguous megablast - сходные последовательности, которые могут являться близкими гомологами (но оба алгоритма производят поиск по паттерну длиной 11 нуклеотидов). Данный пример как раз это иллюстрирует: blastn нашел сходные участки, но они совершенно точно не могут быть гомологами исходной последовательности.
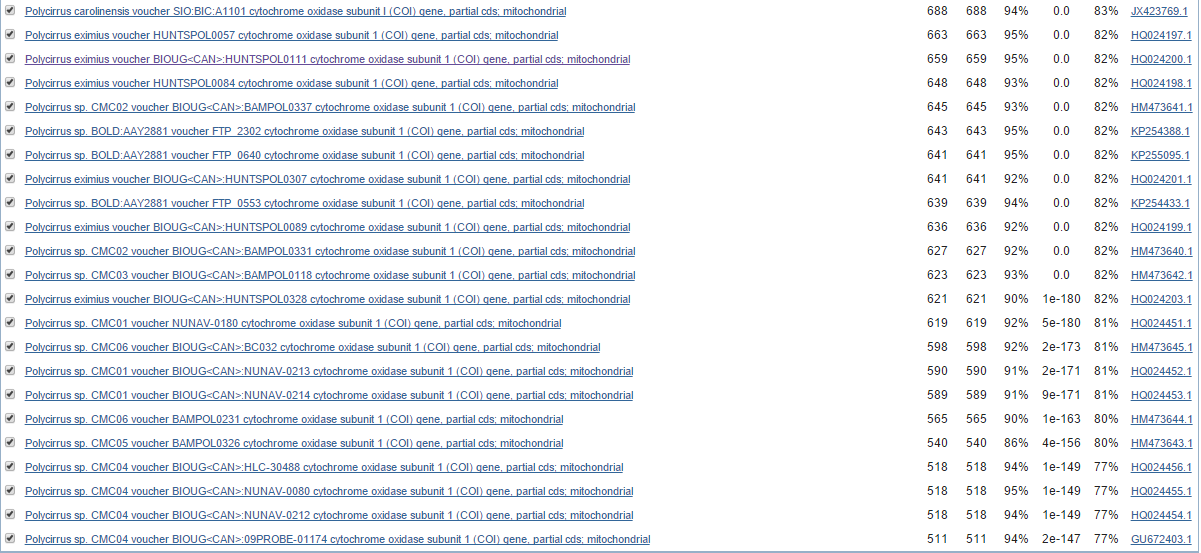

Можно заключить:
- blastn ищет сходные участки любого размера, то есть покрытие исходной последовательности не имеет значения. Используется для поиска ЛЮБЫХ сходных последовательностей.
- discontiguous megablast ищет сходные последовательности, потенциально (!!!) являющиеся близкими гомологами (не можем по выдаче blast судить о гомологии).
- megablast ищет сходные последовательности; по всей видимости, помимо хорошего значения E-value, важен процент сходства (не менее ≈ 90% в моем случае). Можно использовать для поиска очень близких гомологов.
Задание 3 (3.2). Проверка наличия гомологов 5 белков в геноме организма.
В данном задании нужно было проверить наличие гомологов 5 белков в организме Amoboaphelidium (ссылка на сборку генома).
Для этого требовалось провести локальный blast (standalone blast) аминокислотной последовательности каждого из белков против трансляции нуклеотидного банка данных (последовательности генома Amoboaphelidium) в 6 рамках считывания - алгоритм tblastn.
Требовалось отобрать 5 белков, гомологи которых, предположительно, должны встречаться у всех эукариот. В Таблице 2 перечислены выбранные мной белки и кратко указаны их функции.
Таблица 1. Сравнение 3 алгоритмов BLAST.
| Название белка | Имя записи в UniProt (Entry Name) | Идентификатор и ссылка на страницу в UniProt | Функция |
| Гистон H4 | H4_HUMAN | P62805 | Является компонентом нуклеосом, которые наматывают на себя ДНК и компактизуют хроматин, органичивая доступ к ДНК клеточным комплексам, использующим ДНК в качестве матрицы. Таким образом, гистоны играют ключевую роль в транскрипции, репликации и репарации ДНК, а также поддержании стабильности хромосомы. Доступ к ДНК регулируется посредством модификаций гистонов (так называемый гистоновый код). |
| Малая субъединица (35 кДа) фактора сплайсинга U2AF | U2AF1_HUMAN | Q01081 | РНК-связывающий белок, выполняющий функцию фактора сплайсинга пре-мРНК (первичных транскриптов); связывается с динуклеотидом AG в 3'-сайте сплайсинга, "приводит" мяРНП (малую ядрышковую рибонуклеопротеидную частицу) U2 к точке разветвления. Таким образом, является посредником во взаимодействиях, необходимых для точного определения 3'-сайта сплайсинга, а также может играть роль "моста" между энхансерным комплексом и U2AF2, чтобы привлечь последний к следующему интрону. |
| Субъединица 1 фактора инициации транскрипции IIA | TF2AA_HUMAN | P52655 | Является компонентом транскрипционного комплекса РНК-полимеразы II и играет важную роль в активации транскрипции. |
| Каталитическая субъединица ДНК-полимеразы альфа | DPOLA_HUMAN | P09884 | Играет ключевую роль в инициации репликации ДНК. В течение S-фазы клеточного цикла комплекс ДНК-полимеразы альфа (состоящий из каталитической субъединицы POLA1/p180, регуляторной субъединицы POLA2/p70 и двух субъединиц - праймаз PRIM1/p49 и PRIM2/p58) ассоциируется с ДНК в области репликативных вилок через взаимодействия с MCM10 и WDHD1. Праймаза инициирует синтез ДНК, образуя короткие РНК-праймеры на лидирующей и остающей цепях. Эти праймеры исходно удлиняются каталитической субъединицей полимеразы альфа и впоследствии переносятся на полимеразы дельта и эпсилон для процессивного синтеза ДНК на отстающей и лидирующей цепях, соответственно. Перенос происходит по причине того, что полимераза альфа обладает ограниченной процессивностью и не обладает 3'-экзонуклеазной активностью, позволяющей корректировать ошибки, поэтому полимераза альфа не подходит для репликации длинных участков. |
| ДНК-направленная субчастица RPB1 РНК-полимеразы II | RPB1_HUMAN | P24928 | ДНК-зависимая РНК-полимераза II катализирует транскрипцию ДНК в РНК с использованием рибонуклеозидтрифосфатов в качестве субстрата. Данная субъединица - самый крупный каталитический участок РНК-полимеразы II, который синтезирует предшественников мРНК и многие функциональные некодирующие РНК; в комплексе с некоторыми другими субчастицами образует ДНК-связывающий домен - бороздку, в которой синтезируется РНК по матрице ДНК. Содержит С-концевой домен, состоящий из 52 повторов длиной 7 аминокислот (YSPTSPS), который необходим для полимеразной активности [1]. |
Последовательности всех белков я собрала в один fasta-файл, так как это позволяет ускорить процесс работы алгоритма tblastn: fasta.
Затем было необходимо создать банк данных из последовательности генома организма Amoboaphelidium.
Команда: makeblastdb -in X5.fasta -dbtype nucl
Далее я непосредственно запустила tblastn.
Команда: tblastn -query h4.fasta -db X5.fasta -out h4.out -outfmt 7
Для каждого белка программа обнаружила разное (но главное, что ненулевое) число находок. С результатами работы программы можно ознакомиться в файле.
Также я отразила данные о находках в Таблице 3.
Примечание: проанализировав все параметры выдачи программы, я условно определила, что, начиная с E-value=0.1 и выше, находки не будут считаться значимыми (то есть не могут являться потенциальными гомологами. Ситуация для каждого белка неодинакова, но для удобства такую верхнюю границу можно принять. По остальным параметрам не удалось выявить общий критерий, ситуация слишком индивидуальна.
Таблица 3. Данные о находках алгоритма tblastn.
| Название белка (Entry Name) | Число хороших находок в геноме/Общее чило находок | В какой записи лучшая находка | E-value лучшей находки | Процент идентичности лучшей находки | Процент покрытия входной последовательности лучшей находкой | Иллюстрация выдачи tblastn |
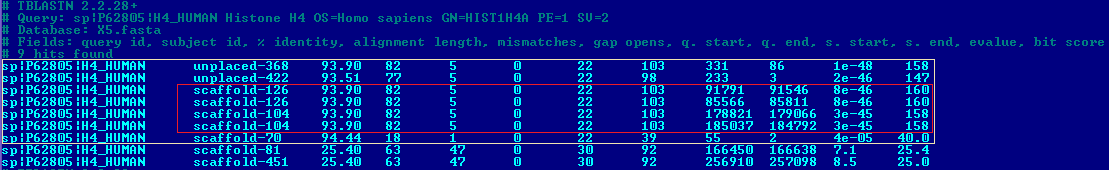
| H4_HUMAN | 7/9 | unplaced-368 | 1e-48 | 93.90 | 79.6 | Рис. 3а |
| U2AF1_HUMAN | 1/4 | unplaced-986 | 1e-46 | 46.67 | 81.25 | Рис. 3b |
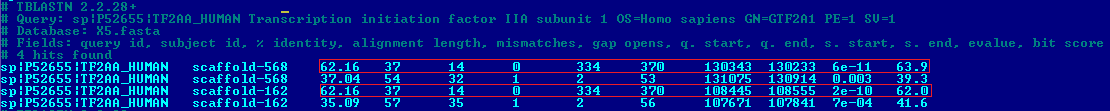
| TF2AA_HUMAN | 4/4 | scaffold-568
scaffold-162 |
6e-11
2e-10 |
62.16 | 9.8 | Рис. 3c |
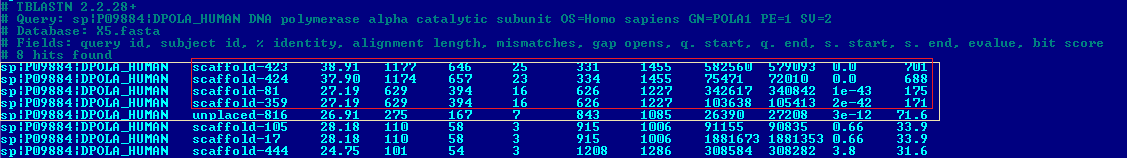
| DPOLA_HUMAN | 5/8 | scaffold-423 | 0.0 | 38.91 | 80.5 | Рис. 3d |
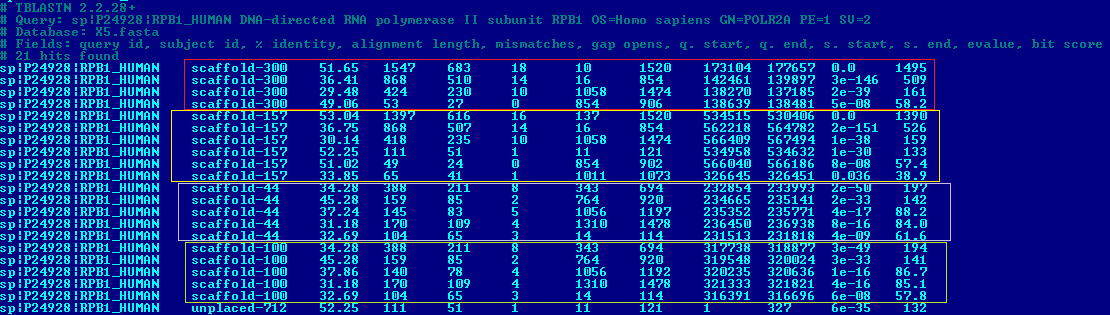
| RPB1_HUMAN | 21/21 | scaffold-157 | 0.0 | 53.04 | 70.9 | Рис. 3e |

Отдельно хочу прокомментировать результаты:
- Для белка H4_HUMAN все хорошие находки имеют крайне сходные параметры (3-6 и вовсе одинаковые, а 7 просто заметно короче). Это позволяет предположить, что мы имеем дело с повторами, причем находки 3-4 и 5-6 даже представляют собой повторы в пределах одного скэффолда. Наверное, это можно объяснить значимостью гистона Н4 в жизнеспособности организма; возможно, он представлен в нескольких копиях для уменьшения вероятности гибели организма при нарушении гена.
- Для белка U2AF1_HUMAN я признала хорошей только одну находку, так как она намного лучше остальных по всем параметрам. Однако все остальные находки имеют крайне сходные параметры, что настораживает, но мне здесь не хватает знаний, чтобы это прокомментировать. ((
- Для белка TF2AA_HUMAN представлено 2 "сета" хороших находок, каждый из которых относится к своему скэффолду. Параметры находок 1, 3 и 2, 4 имеют крайне высокое сходство. Итак, рискну предположить, что в каждом из двух скэффолдов содержится по копии гена, при этом в каждом скэффолде присутствует пара находок, которые могут соответствовать двум экзонам, разделенным интроном (организм же эукариотический!), что подтверждается соответствием разным участкам входной последовательности и банка данных.
- Очень интересная ситуация с белком DPOLA_HUMAN: из 5 хороших находок первая и вторая пары также имеют очень схожие значения параметров между собой. Также пары отличаются друг от друга длиной выравнивания, но вряд ли здесь можно предположить разделение находок на экзоны, так как один участок входной последовательности полностью включает в себя другой. Еще одна интересная деталь: в первой паре находок гены белка направлены в одну сторону (лежат на одной цепи), а во второй паре гены направлены в разные стороны (лежат на разных цепях).
- Для белка RPB1_HUMAN я признала все находки хорошими. Находки относятся к 4 скэффолдам, причем находки скэфолдов 300 и 157 и 44 и 100 имеют между собой сходные значения параметров. В итоге можно предположить, что ген представлен в нескольких (2?) копиях, находки внутри каждой пары скэффолдов относятся к разным копиям, находки каждой пары скэффолдов, предположительно, соответствуют экзонам, разделенным интронами. Находки внутри каждого скэффолда так же могут соответствовать экзонам.
Замечания:
- Локальный blast не выдает информации о покрытии исходной последовательности. Это крайне неудобно, для подсчета пришлось делить длину выравнивания на длину соответствующего белка.
- Я слукавила: была попытка найти гомолог центромерного белка С, но blast не нашел ни одной находки, что мне показалось очень странным, так как это крайне важный белок
эукариот (как минимум, я знала, что он гомологичен у дрожжей и высших эукариот). Было предположение, что у организма эту функцию все-таки выполняет негомологичный белок
(это было бы возможно в случае принадлежности организма какому-нибудь экзотическому таксону).
Использованные ресурсы:- [1] - РНК-полимераза II