Ферменты и метаболические пути. База данных KEGG
Работа с базой данных KEGG ORTHOLOGY
В данном задании требовалось проверить, являются ли члены разных ортологических рядов KEGG гомологичными белками, и проанализировать их филогенетические отношения.
Для выполнения задания я взяла реакцию гидролиза фосфо-эфирной связи (превращения 1D-мио-инозитол-1,4-бисфосфата в 1D-мио-инозитол-4-фосфат и 1D-мио-инозитол-1,3,4-трифосфата в 1D-мио-инозитол-3,4-бисфосфат) (EC: 3.1.3.57) из метаболического пути биосинтеза инозитолфосфата (выделена голубым цветом на Рис. 1).
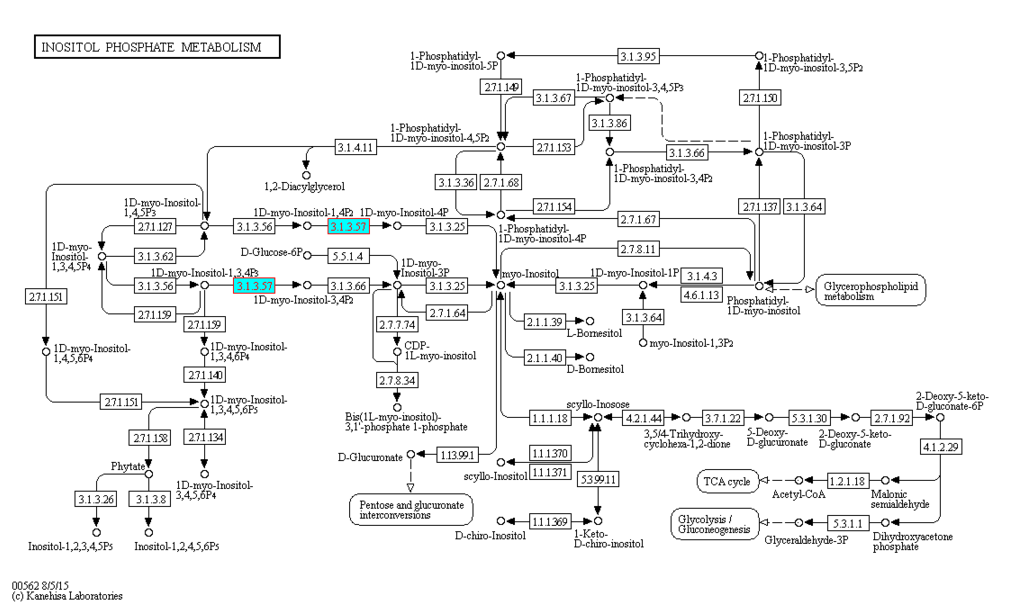
Данную реакцию катализируют два ортологических ряда инозитол-полифосфат-1-фосфатаз (идентификаторы и ссылки на записи в БД KEGG ORTHOLOGY представлены в Таблице 1).
Таблица 1. Ортологические ряды белков, катализирующие реакцию 3.1.3.57.
| Идентификатор в БД KEGG ORTHOLOGY | Количество белков в ряду | Ссылка на запись в БД KEGG ORTHOLOGY |
| K01107 | 62 | KO |
| K15422 | 63 | KO |
Затем были послучены последовательности белков с помощью сервиса "Retrieve/ID mapping" из БД Uniprot. Идентификаторы преобразованы следующим образом: ХХХХХХ_ХХХХХ|K*****, где К***** - идентификатор ортологического ряда в БД KEGG ORTHOLOGY.
Полученные последовательности были выровнены с помощью программы Muscle (выравнивание в формате fasta).
Было выполнено множественное выравнивание последовательностей Выравнивание было открыто в JalView (ссылка на проект).
Гомологичность белков в выравнивании
В прикрепленном выше проекте исходное выравнивание представлено в окне aligned, раскраска ClustalX. Принятие решения о том, являются ли белки разных ортологических рядов гомологами, является в высшей степени нетривиальной задачей, так как нет четкого критерия.
В выравнивании видно, что довольно много "хороших" консервативных позиций, особенно если учесть небольшую длину белков (280-420 остатков) - для таких коротких белков это вообще неплохо. В пользу гомологичности также свидетельствует тот факт, что среди этих позиций присутствуют абсолютно консервативные, причем они часто представлены гидрофильными аминокислотами. Присутствуют, однако, колонки, в которых остаток четко определяется принадлежностью к ортологическому ряду, но их не так много (определено посредством нетренированного глазомера). В случае функционально консервативных позиций часто остатки "перемешаны" между всеми последовательностями, а не в пределах кластеров, что тоже говорит в поддержку гомологичности. В невыровненных участках остатки также разбросаны не только между кластерами, но и внутри них.
Нельзя не заметить, что кластеры чисто визуально различимы по выравниванию, но это по большей части происходит из-за нескольких вставок. Многие гэпы связаны с определенными короткими белками, которые будут устранены позднее.
Вердикт: множественное выравнивание существует, так как выровнялось много консервативных позиций, а различия, где таковые имеются, чаще наблюдаются между всеми последовательностями, а не между кластерами.
Проверка выравнивания
- Белков, очень плохо выровненных с остальными и составляющих отдельный кластер, обнаружено не было.
- В обоих рядах встретились короткие белки, содержащие много гэпов в тех колонках, где в других последовательностях выравнивания консервативные колонки.
Их потребовалось удалить: V4SX75_9ROSI|K15422 (115), V4T781_9ROSI|K15422 (179), Q6PB10_XENLA|K01107 (338), C1N5L7_MICPC|K15422 (349), I1E8Q6_AMPQE|K01107 (299),
D8U9Y8_VOLCA|K15422 (431). Выравнивание с удаленными коротками белками представлено в проекте JalView (окно clean).
Оставшиеся в выравнивании последовательности (fasta) были использованы для построения филогенетического дерева методом Neighbor-Joining со 100 бутстреп-репликами (программа MEGA). Результат на Рис. 2.

Рис. 2. Филогенетическое дерево отобранных последовательностей, построенное методом Neighbor-Joining со 100 бутстреп-репликами (программа MEGA).
Анализ филогенетического дерева:
- Дерево четко распадается на клады, соответствующие отдельным ортологическим рядам. Причем бутстреп-поддержка ветви, разделяющей дерево на эти клады, равна 99, что свидетельствует о достоверности этого разделения.
- Что касается длин ветвей, то большие группы белков имеют приблизительно одинаковые длины ветвей, между группами расстояние может немного отличаться. Однако есть и пример ветви, сильно выбивающейся среди остальных - A4HJT1_LEIBR|K01107. Если обратиться к выравниванию, то можно заметить, что она действительно выровнена хуже остальных. Но при первичном рассмотрении (пункт 1 проверки выравнивания) мне не удалось ее выделить среди остальных.