
Для построения дерева из базы полных геномов NCBI были взяты последовательности 16S РНК тех же бактерий, что и в первом практикуме и объединены в один файл.
Далее последовательности былит выровнены в программе Jalview:
Выравнивание 16s РНК последовательностей бактерий
Данное выравнивание было открыто в программе MEGA и методом Maximum likelihood было получено дерево:
| Дерево, полученное из выравнивания 16s РНК последовательностей | Эталонное дерево |
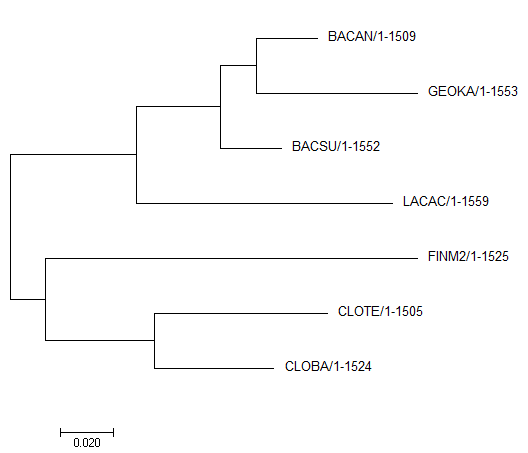 | 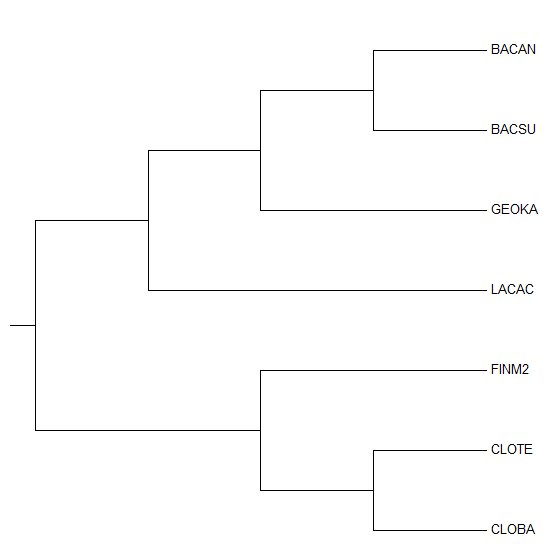 |
Как видно, топология дерева почти полностью совпадает с эталонным: отличаются только ветвь {BACAN, GEOKA} vs {BACSU, LACAC, FINM2, CLOTE, CLOBA}
(у эталонного же {BACAN, BASCU} vs {GEOKA, LACAC, FINM2, CLOTE, CLOBA}).
Если дерево, построенное по нуклеотидным последовательностям, сравнивать с деревьями,
построенными по белковым, то вторые существенно проигрывают по совпадению с эталонным.
Чтобы найти все гомологи белка CLPX_BACSU, я слила все протеомы бактерий из базы данных Uniprot
(P:\y15\term4\Proteomes) в один файл proteomes.fasta командой cat.
Далее была создана даза данных для blastp
командой
makeblastdb -in proteomes.fasta -out db.fasta -dbtype protЗатем был осуществлен поиск гомологов:
blastp -query CLPX_BACSU.fasta -db db.fasta -evalue 0.001 -out hmlg.txtВ результате был получен файл hmlg.txt cо списком из 32 гомологов.
Список гомологов, полученный с помощью алгоритма blastp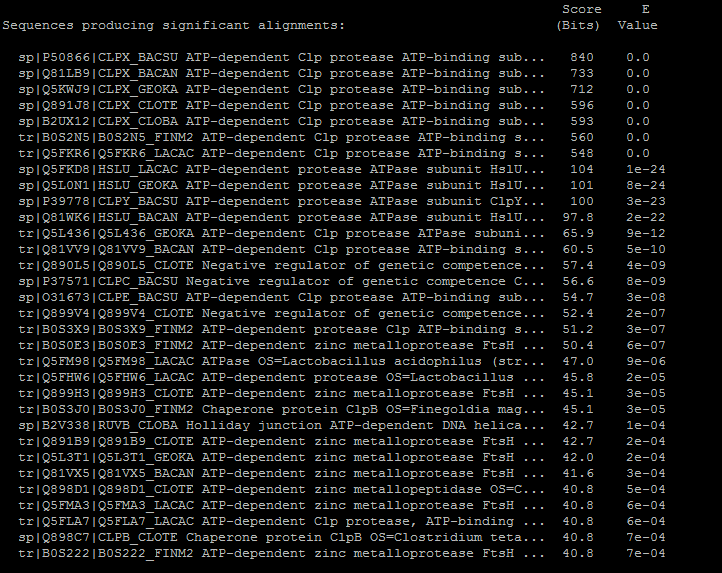
С помощью команды fetch в программе Jalview по мнемоникам из полученного файла были скачаны последовательности и
выровнены алгоритмом Muscle.
Файл с итоговым выравниванием был открыт в программе MEGA.
Затем методом Neighbour-joining было построено филогенетическое дерево.
Филогенетическое дерево гомологов белка CLPX_BACSU
На изображении зеленым отмечены примеры паралогов, синим - ортологов (соответствующие цифры обозначают группы, в которых все белки попарно ортологи или паралоги), рамками отмечены примеры эволюционных событий: красным - видообразование, фиолетовым - дупликация.
© Кучеренко Варвара 2015