EMBOSS
Упражнения
| Опция | Что делает | Команда | Результат |
| seqret | Собирает несколько файлов в fasta-формате в единый файл. Например, здесь я привожу команду, собирающую все файлы записи Swissprot, начинающиеся на 01: | 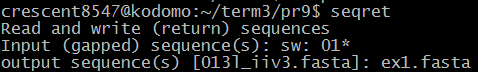 |
ex1 |
| seqretsplit | Один файл в формате fasta делится на несколько. Разделим все файлы записи Swissprot, заканчивающиеся на R4: | 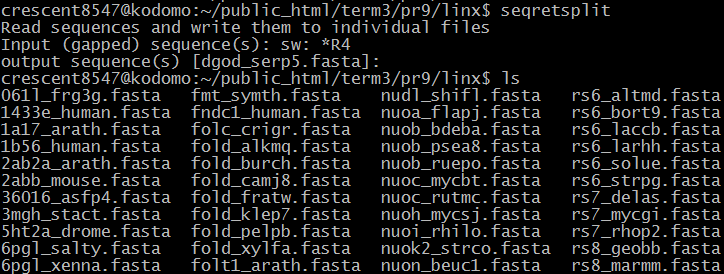 |
(см. рис.2) |
| transeq | Транслирует последовательность нуклеотидов в последовательность аминокислот. На вход был подан файл ex3in.fasta,
содержащий последовательность гена алкогольдегидрогеназы организма Arthrobacter sp. |
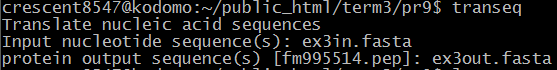 |
ex3 |
| seqret(2) | Так же способен перевести выравнивание из fasta-формата в формат msf. На вход было взято выравнивание | 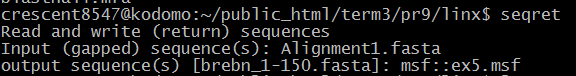 |
ex4 |
| cusp | Ищет частоты кодонов в кодирующей последовательности (например, seq5.fasta) и записывает их в таблицу; также выдает частоту GC в последовательности и соответствие кодонов аминокислотам. | 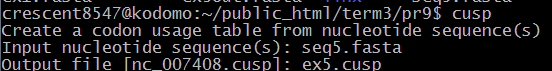 |
ex5.cusp |
| featcopy | Переводит переводит аннотации особенностей в записи gb-формата в табличный формат .gff. | 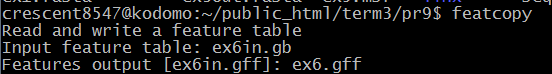 |
ex6 |
| extractfeat | Из файла последовательности гена получаем кодирущие последовательности. | 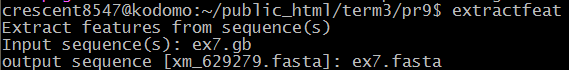 |
ex7 |
| transeq(2) | Транслирует данную нуклеотидную последовательность в шести рамках. На вход берем файл ex3in.fasta. | transeq ex3in.fasta ex8.fasta -frame 6 | ex8 |
| compseq | Считает структуру уникальных слов (например, динуклеотидов, как в задании) в последовательности. На вход берем последовательность гена из упражнения 7. | 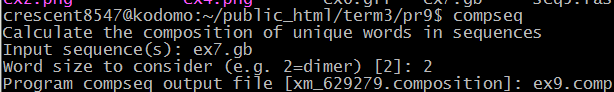 |
ex9.comp |
| shuffle | Перемешивает буквы в данной нуклеотидной последовательности. На вход берем нуклеотидную последовательность предсказанного белка
Dictyostelium discoideum AX4 hypothetical protein (helA) mRNA; по полученной "перемешанной" последовательности был запущен поиск blastn с параметрами по умолчанию; В результате получили 172 находки,
из них 43 "достоверных", т.е. с E-value меньше 0.1. Разумеется, к исходной последовательности они отношения не имеют: Query cover у всех не превышает 3%: |
shuffle -o ex10out.fasta ex10.fasta | ex10 |
Задания: сравнение аннотации генов белков в одной хромосоме бактерии или археи с трансляциями длинных открытых рамок считывания.
Бактерию взял "свою" из прошлого семестра, Bacteroides thetaiotaomicron, штамм VPI-5482. Подробнее почитать про сам организм можно, например, здесь.
Бактерия имеет одну кольцевую хромосому: Последовательность хромосомы в формате GenBank.
AC записи: NC_004663
Последовательность в fasta-формате: ссылка.
1. Получение трансляции открытых рамок с помощью команды getorf пакета EMBOSS.
Программа getorf извлекает открытые рамки считывания только с нуклеотидных последовательностей (а их в формате .gb нет), поэтому была использована запись в fasta-формате. Так же были прописаны следующие опции:
- Таблица генетического кода для данного генома: -table 11
- Минимальная длина открытой рамки: -minsize 180
- Кольцевая хромосома: -circular
- Трансляции открытых рамок от стоп кодона до стоп кодона: -find 0

Результат - файл с открытыми рамками считывания.
2. Получение списка координат и ориентаций найденных открытых рамок с помощью infoseq.
Команда: infoseq bacteroides.orf -only -name -sprotein1 -length -description > infoseqresult.
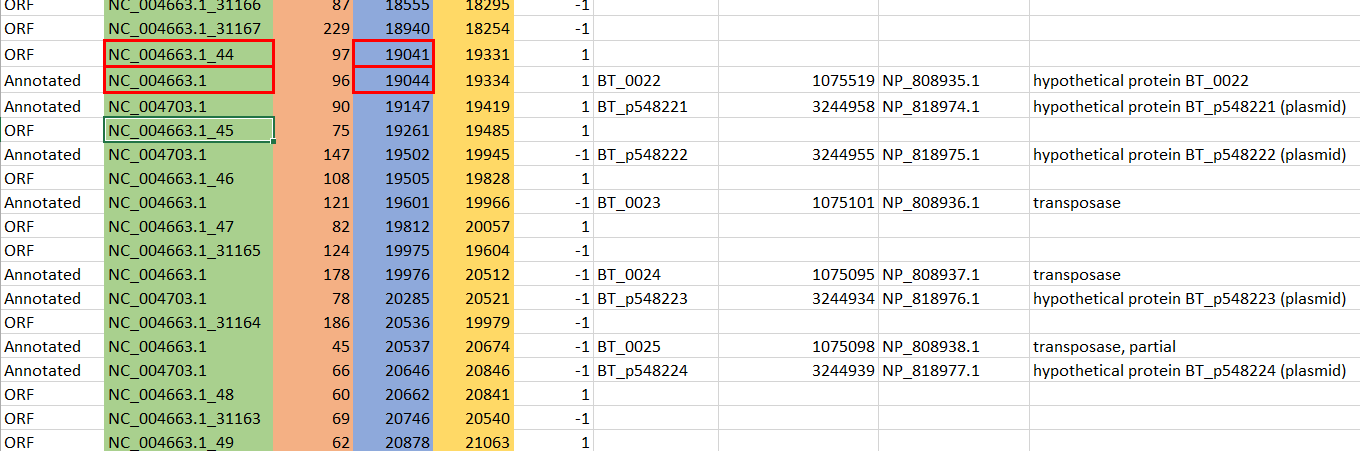
Полученный файл был отредактирован с помощью Excel:
Таблица результатов
3. Получение списка аннотированных генов белков.
Была скачана и приведена в соответствующий вид Таблица аннотированных белков Bacteroides thetaiotaomicron. Так же, был получен fasta-файл с последовательностями
всех аннотированных белков, в форме .gz - архива.
После этого две таблицы были объединены в одну общую, и отсортированы по столбцы началу в геноме; Однако начало ни одного из аннотированных генов не совпало
с началом на открытых рамках считывания, в среднем начало генов отличается на несколько десятков нуклеотидов, в лучшем случае - на три (см. рис. 10)
Назад к странице семестров