Далее нужно было создать 3 файла в FASTA-формате, содержащие последовательность моего белка, а также последовательности каждого из соответствующих мутантов:
Далее нужно было найти то место в моем исходном белке, из которого был вырезан мутированный фрагмент, и вручную выровнять его.
Для 1-го мутанта:
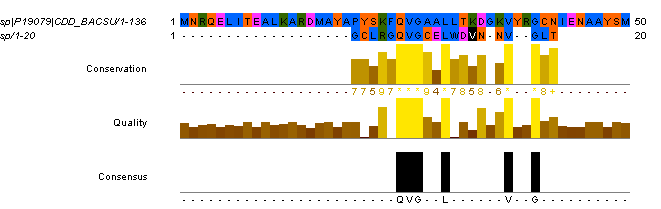
%Сходства=30%
%Совпадения=50%
Для 2-го мутанта:
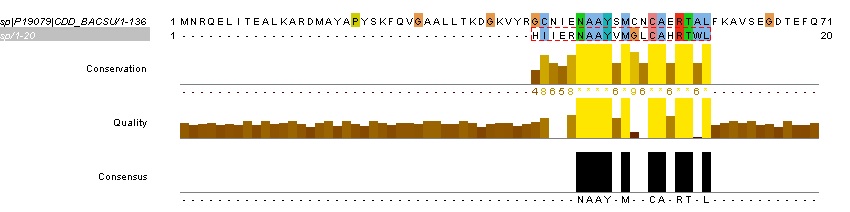
%Сходства=50%
%Совпадения=60%
Для 3-го мутанта:
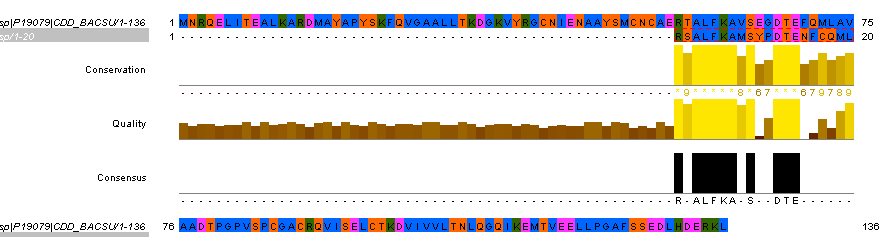
%Сходства=50%
%Совпадения=75%
Данное выравнивание сохранено в файле alignment1.msf.
Процент сходства- 43%
Процент идентичности- 33%
2.Нужно построить выравнивание последовательностей моего белка и его предполагаемых ортологов или гомологов (всего 3 последоватетельности), найденных на предыдущих занятиях.
Гомологи моего белка представлены в этом файле- homologs.fasta
Далее выравниваем их с помощью программы Muscle
Получаем три выравнивания:
Далее, используя команду infoalign, мы получаем:
Сохраненный мной проект Jalview:
© Прозоров Данила