| Главная страница | Семестры | О себе | Ссылки |


Получение консенсусной последовательности по двух хроматограммам для комплиментрных цепей
В данном практикуме необходимо было проанализировать и отредактировать последовательности, которые были получены из хроматограмм, выданных капиллярным секвенатором по Сэнгеру.
Для работы были взяты файлы хроматограмм, выданные секвенатором на ББС МГУ, в формате .ab1 (прямая цепь, обратная цепь).
Для обработки результатов секвенирования использовалась программа Chromas Lite. Результаты работы в ней:
Выравнивание прямой и комплементарной к обратной последовательности с отмеченными измененными нуклеотидами в проекте JalView. (ссылка)
Файл в формате .fasta с "чистой" прочтенной последовательностью. (ссылка)
Ход работы
Я открыла два окна Chromas Lite: одно с прямой последовательностью, другое - с комплементарной к обратной (получена с помощью опции Reverse+Complement). Для дальнейшего сравнения двух последовательностей требовалось удалить нечитаемые области с 5' и 3' концов. Это (также представлено на рис. 1):
3' прямой - 1-18 позиции.
5' прямой - 377-380 позиции.
3' комплиментарной обратной - 1-5 позиции.
5' комплиментарной обратной - 347-380 позиции.
Не все из позиций, отмеченных мной как нечитаемые, были помечены в файле знаком N. Были смазанные пики (как, например, 347 позиция цепи, комплиментарной обратной), наложенные пики (к примеру, 378 позиция прямой цепи).
|
|
3'-конец прямой цепи |
5'-конец прямой цепи |
|
|
3'-конец цепи, обратной комплиментарной |
5'-конец цепи, обратной комплиментарной |

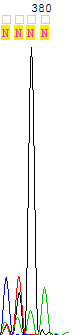
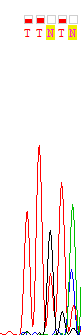
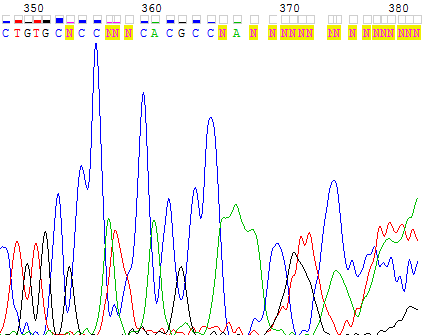
Рис. 1. Нечитаемые области
Нечитаемые области были удалены. О самих хроматограммах я могу сказать, что они кажутся мне качественными. Самые низкие пики не более, чем в 4 раза, выше самых низких. Также заметно, что пики гуанинов на прямой цепи (соответственнло, цитозинов на цепи, комплиментарной обратной) самые высокие. Пики аденинов на прямой цепи тоже часто выделяются, но не так ярко, как у гуанинов.
Затем я приступила к редактированию цепей. Каждое редактирование приведено на рисунках и описано ниже. Характерной особенностью моей хроматограммы было большое количество наложенных пиков. Многие из них были определены программой как N, хотя отчетливо видно только два пика, притом на обеих хроматограммах. Имею смелость предположить, что такие двоиные пики соответствуют полиморфизмам в исходной ДНК.
|
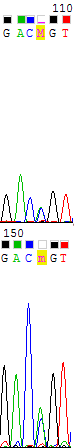
1) N на 51 позиции прямой цепи уточнено до m по другой цепи. Два наложенных пика хорошо различимы на обеих хроматограммах. 2) N на 60 позиции прямой цепи аменен на g по данным из другой хроматограммы. Да даже и без дополнительных данных пик гуанина хорошо проступает на фоне скачка шума. 3) N на 72 прямой и N на 116 комплиментарной обратной позициях я заменила на w, так как своими глазами вижу наложенные пики аденина и тимина. |
Рис. 2. Примеры исправлений хроматограммы |
|
|
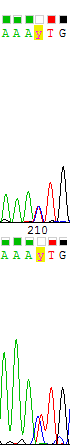
4) N 152 позиции цепи, обратной комплиментарной, заменено на m, потому что два наложенных пика ясно видны, но не были распознаны программой. 5) N на 166 позиции прямой и 210 комплиментарной обратной заменены на y на основании очевидности двух пиков в обоих случаях. |
Рис. 3. Примеры исправлений хроматограммы |
|
|
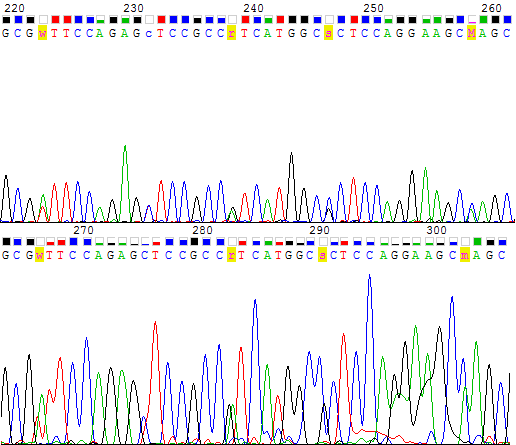
6) N на 222 позиции прямой и 266 комплиментарной обратной заменены на w из видимости парных пиков на хроматограммах. 7) N на 231 месте первой последовательности заменен на C на основании данных второй последовательности и внешнего вида хроматограммы. 8) N на 238 позиции прямой и 282 комплиментарной обратной заменены на r, потому что так видно по хроматограмме. 9) N на 246 позиции прямой и 290 комплиментарной обратной заменены на s. Двойной пик наблюдается из хроматограммы. Однако если бы я не видела хроматограмму прямой цепи, я бы сомневалась насчет комплимента обратной: вставлять в последовательность еще один нуклеотид или предполагать полиморфизм? 10) N на 302 позиции комплимента обратной заменено на m. Парный пик виден достаточно хорошо. |
Рис. 4. Примеры исправлений хроматограммы |
|
 .
. 
Далее последовательности были вручную выровнены в JalView (результат по ссылке выше). Получилось, что несовпадающих нуклеотидов нет. По выравниванию составлена "чистая" последовательность, также выложенная в начале практикума.
Выяснение данных о последовательности методом BLASTN
Для выяснения, что за ДНК была отсеквенирована и какому организму она принадлежала, я использовала BLASTN NCBI. Ввела консенсусную последовательность, запустила blast. Верхние результаты представлены на рис. 5.
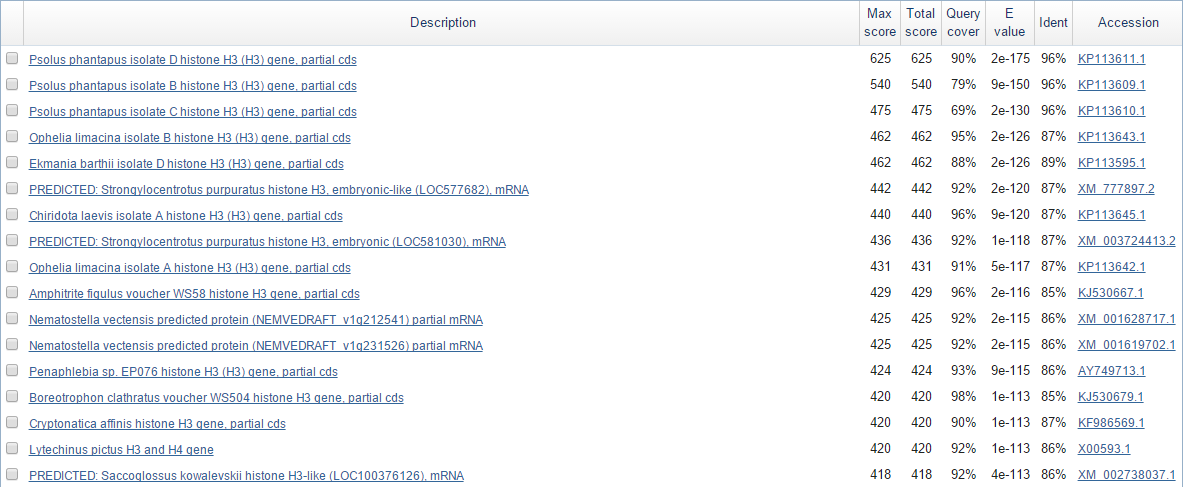
Рис. 5. Лучшие результаты работы blastn по консенсусной последовательности
E value всех представленных на рис. 5 находок почти ноль, значит, они не случайны, на их основании можно делать выводы о моей последовательности. А выводы напрашиваются такие:
Все последовательности, за исключением двух неопределенных, относятся к гистону H3. С очень высокой долей вероятности моя последовательность тоже кодирует какую-то часть этого белка.
Лучшие находки относятся преимущественно к разным, далеким друг от друга, типам царства Metazoa: Echinodermata, Hemichordata, Lophotrochozoa, Cnidaria. Все это морские беспозвоночные животные. Однозначно, что мое животное - морское беспозвоночное.
Гистон H3 встречается если не у всех, то у абсолютного большинства эукариот и выполняет во всех организмах очень сходную функцию (упоковка ДНК в нуклеосомы). Наверняка последовательность ДНК, его кодирующая, очень консервативна и будет сходна даже у таксономически далеких друг от друга эукариотических организмов. Потому я не возьмусь судить о том, к какому из "мелких" таксонов принадлежит мой организм, по последовательности H3.
Самые близкие к моей последовательности (перекрывание 96%) принадлежат морскому огурцу Psolus phantapus. Но исходя из предыдущего пункта и из того, что ненамного менее сходные с моей последовательности принадлежат к организмам, отличающимся от морского огурца на уровне типа, я не могу утверждать, что мое животное - Psolus phantapus.
4 из 5 организмов, чьи последовательности ближе всего к моей, принадлежат к подтипу Echinozoa. Это означает, что мое животное, скорее всего, принадлежит к этому подтипу.
Также я построила выравнивание моей консенсусной последовательности с тремя находками с наибольшим перекрыванием и из разных организмов.
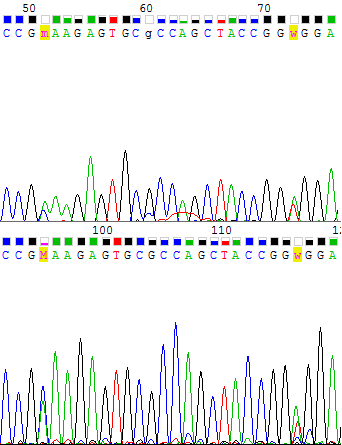
Нечитаемые хроматограммы
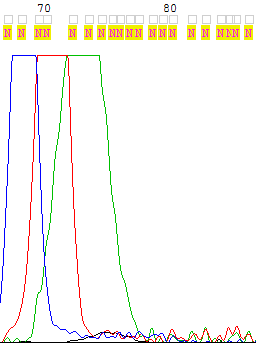
Рис. 6. Участок нечитаемой хроматограммы
В качестве примера плохой хроматограммы я скачала WS2943_SP6R.ab1. Ни один нуклеотид не определен (3 нуклеотида выделены программой, но это однозначно случайно), шум неотличим от сигнала. Уровень сигнала в разных местах хроматограммы отичается на порядки.
© Дарья Горбачева | изменено 16.11.2015 |