Семестр 2. Практикум 12. Алгоритмы и программы множественного выравнивания
Содержание
2. Сравнение выравниваний одних и тех же последовательностей тремя программами
2.1. Последовательности и программы
Для сравнения выбрана доменная архитектура BAR + SH3_1 из практикума 11.
Взяты 7 белков из семейства амфифизин/эндофилин, несущих оба домена:
| UniProt AC | Название | Организм |
|---|---|---|
| P49418 | AMPH_HUMAN — Amphiphysin | H. sapiens |
| O00499 | BIN1_HUMAN — Bridging integrator 1 | H. sapiens |
| Q9UBW5 | BIN2_HUMAN — Bridging integrator 2 | H. sapiens |
| Q99961 | SH3G1_HUMAN — Endophilin-A2 | H. sapiens |
| Q99962 | SH3G2_HUMAN — Endophilin-A1 | H. sapiens |
| Q9Y371 | SHLB1_HUMAN — Endophilin-B1 | H. sapiens |
| P39743 | RV167_YEAST — RVS167 | S. cerevisiae |
Те же последовательности выравнены тремя программами:
- A — MUSCLE (EBI, v3.8), 802 колонки
- B — MAFFT (CBR, режим auto/L-INS-i), 824 колонки
- C — T-Coffee (CRG, regular mode), 858 колонок
FASTA: A (MUSCLE) · B (MAFFT) · C (T-Coffee)
VerAlign на момент подготовки отчета выдавал ошибку "Queue job has crashed", поэтому сравнение выравниваний выполнено программно, с помощью скрипта на Python: для каждой пары выравниваний построено отображение "остаток - колонка" в каждом из них; колонки считаются одинаково выравненными, если все последовательности имеют тот же остаток в той же позиции в обоих выравниваниях.
stdout работы скрипта.
2.2. Сравнение A (MUSCLE) и B (MAFFT)
Совпадающих колонок: 225 из 802 (28,1% от A; 27,3% от B).
Таблица 1. Блоки одинаково выравненных колонок, A vs B (длина ≥ 2)
| (s1, f1) = (s2, f2) | Длина | Примечание |
|---|---|---|
| (242, 282) = (254, 294) | 41 | наибольший блок |
| (660, 696) = (676, 712) | 37 | |
| (16, 50) = (16, 50) | 35 | начало выравнивания, совпадает позиционно |
| (594, 617) = (610, 633) | 24 | |
| (211, 232) = (223, 244) | 22 | |
| (494, 508) = (513, 527) | 15 | |
| (525, 536) = (544, 555) | 12 | |
| (792, 802) = (814, 824) | 11 | конец выравнивания |
| (633, 639) = (649, 655) | 7 | |
| (9, 12) = (9, 12) | 4 | |
| (469, 472) = (488, 491) | 4 | |
| (480, 483) = (499, 502) | 4 | |
| (375, 377) = (396, 398) | 3 | |
| (139, 140) = (153, 154) | 2 | |
| (643, 644) = (659, 660) | 2 |
Одиночные совпадающие колонки вне блоков: (128, 142), (433, 452).
Крупнейшие несовпадающие участки в выравнивании A:
- 697–791 (95 колонок)
- 283–374 (92 колонки)
- 378–468 (91 колонка)
- 51–138 (88 колонок)
- 141–210 (70 колонок)
2.3. Сравнение A (MUSCLE) и C (T-Coffee)
Совпадающих колонок: 213 из 802 (26,6% от A; 24,8% от C).
Таблица 2. Блоки одинаково выравненных колонок, A vs C (длина ≥ 2)
| (s1, f1) = (s2, f2) | Длина | Примечание |
|---|---|---|
| (16, 53) = (16, 53) | 38 | наибольший блок |
| (242, 273) = (258, 289) | 32 | |
| (480, 508) = (511, 539) | 29 | |
| (211, 232) = (227, 248) | 22 | |
| (361, 372) = (382, 393) | 12 | |
| (792, 802) = (848, 858) | 11 | конец выравнивания |
| (525, 534) = (563, 572) | 10 | |
| (660, 668) = (694, 702) | 9 | |
| (671, 679) = (705, 713) | 9 | |
| (633, 639) = (667, 673) | 7 | |
| (278, 282) = (294, 298) | 5 | |
| (10, 12) = (10, 12) | 3 | |
| (375, 377) = (396, 398) | 3 | |
| (470, 472) = (501, 503) | 3 | |
| (735, 737) = (771, 773) | 3 | |
| (105, 106) = (110, 111) | 2 | |
| (139, 140) = (155, 156) | 2 | |
| (314, 315) = (330, 331) | 2 | |
| (327, 328) = (343, 344) | 2 | |
| (355, 356) = (376, 377) | 2 | |
| (358, 359) = (379, 380) | 2 | |
| (442, 443) = (469, 470) | 2 | |
| (445, 446) = (472, 473) | 2 |
Одиночные совпадающие колонки вне блоков: (127, 143).
Крупнейшие несовпадающие участки в выравнивании A:
- 535–632 (98 колонок)
- 141–210 (70 колонок)
- 680–734 (55 колонок)
- 54–104 (51 колонка)
- 738–791 (54 колонки)
2.4. Обсуждение
MAFFT (B) ближе к MUSCLE (A), чем T-Coffee (C): совпадение 28,1% против 26,6%. При этом блоки в паре A–B длиннее (максимальный блок 41 против 38), а число блоков меньше (15 против 23). Это означает, что MAFFT и MUSCLE «соглашаются» в более протяжённых консервативных участках, тогда как T-Coffee дробит совпадения на большее число коротких блоков[1].
Проект Jalview (выравнивания A, B, C)
3. Структурное выравнивание и сравнение с выравниванием MSA
3.1. Выбор структур
Выбраны три структуры BAR-домена из PDB — все принадлежат семейству амфифизин/эндофилин:
| PDB | Белок | Организм | Длина |
|---|---|---|---|
| 1X03:A | Endophilin-A1 BAR domain (SH3G2) | H. sapiens | 210 а.о. |
| 1URU:A | Amphiphysin BAR domain | D. melanogaster | 217 а.о. |
| 4ATM:A | Amphiphysin BAR domain | H. sapiens | 221 а.о. |
3.2. Визуализация совмещения структур
Визуализация совмещения реализована в PyMOL и транслирована в PDB для отображения в Mol*.
Для отображения совмещенных структур в Mol* понадобилось дополнительно изменить идентификаторы цепей в PyMOL и сохранить в виде нового PDB.
fetch 1x03 fetch 1uru fetch 4atm super 1uru, 1x03 super 4atm, 1x03 color red, 1x03 color blue, 1uru color green, 4atm alter 1uru, chain="B" alter 4atm, chain="C" sort save prakt-D-superposed.pdb, 1x03 or 1uru or 4atm
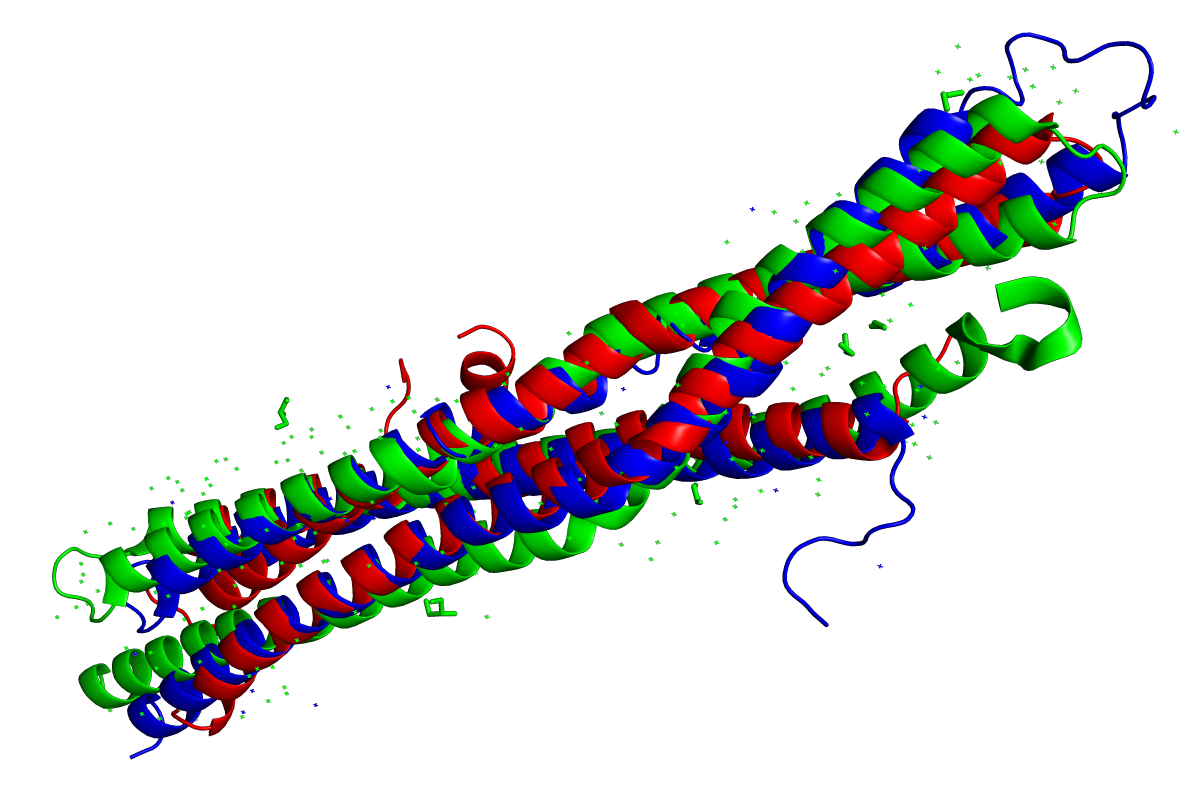
Рисунок 1. Совмещение 1X03, 1URU, 4ATM.
- Красный —
1X03(Endophilin-A1, Human) - Синий —
1URU(Amphiphysin, Drosophila) - Зелёный —
4ATM(Amphiphysin, Human)
3D-модель 1. Совмещение 1X03, 1URU, 4ATM.
3.3. Сравнение структурного выравнивания с MSA
Множественное структурное выравнивание последовательностей (259 колонок) получено с помощью сервиса PDBeFold (Secondary Structure Matching, режим Multiple). Результат выравнен MUSCLE (265 колонок). Сравнение двух выравниваний выполнено программно скриптом из задания 2.
stdout работы скрипта.
FASTA: структурное (PDBeFold) · MSA (MUSCLE)
Совпадающих колонок: 8 из 259 (3,1% от структурного; 3,0% от MSA).
Таблица 3. Блоки одинаково выравненных колонок, структурное (s1) vs MUSCLE (s2)
| (s1, f1) = (s2, f2) | Длина | Примечание |
|---|---|---|
| (1, 3) = (1, 3) | 3 | начало выравнивания |
| (62, 63) = (84, 85) | 2 | |
| (172, 173) = (178, 179) | 2 |
Одиночные совпадающие колонки вне блоков: (78, 96).
Крупнейшие несовпадающие участки в структурном выравнивании:
- 4–61 (58 колонок)
- 64–171 (108 колонок) — наибольший
- 174–259 (86 колонок)
3.4. Обсуждение
Совпадение структурного и последовательностного выравниваний крайне низкое — всего 3,1%. Это закономерный результат: последовательностная идентичность между Endophilin-A1 и Amphiphysin составляет около 10%, что ниже «порога Дулитла» (~25%), при котором последовательностное выравнивание становится ненадёжным.
PDBeFold выравнивает структуры по геометрии Cα-атомов и выявляет консервативную пространственную организацию там, где MUSCLE «не видит» сходства в последовательности[2]. Все три структуры образуют одинаковую серповидную форму, однако аминокислоты, занимающие одинаковое положение в пространстве, часто оказываются различными — именно поэтому большинство структурно выравненных пар в последовательностном выравнивании расположены в разных колонках.
Три небольших совпадающих блока (суммарно 7 колонок) соответствуют наиболее консервативным участкам ядра BAR-домена, которые сохранили как пространственную геометрию, так и частичную последовательностную сходимость. Это подтверждает, что структура консервативнее последовательности — характерная особенность отдалённо гомологичных доменов[3].
Проект Jalview (структурное + MSA)
4. Список литературы
- Edgar, R.C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. doi: 10.1093/nar/gkh340
- Peter, B.J., Kent, H.M., Mills, I.G., Vallis, Y., Butler, P.J.G., Evans, P.R., McMahon, H.T. (2004). BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science, 303(5657), 495–499. doi: 10.1126/science.1092586
- Chothia, C., Lesk, A.M. (1986). The relation between the divergence of sequence and structure in proteins. EMBO Journal, 5(4), 823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x
5. Благодарности
Выражаю благодарность Гончарову Евгению Борисовичу за помощь в разработке и тестировании скрипта для сравнения выравниваний.