Вернуться на страницу семестра
Задача: картировать чтения, полученные в результате секвенирования транскриптома (человек, версия сборки генома hg19)
Часть I: подготовка чтений
0. Создание рабочей директории.
В директории /nfs/srv/databases/ngs/ создана директория e.mironova (ещё в предыдущей работе), для этой работы в этой директории создана папка pr12.
В неё скопированы файлы с ридами (chr21.1.fastq и chr21.2.fastq - это 2 биологические реплики) и
разметка человеческого генома по версии Gencode19 для сборки hg19. Разметка в формате .gtf лежит на kodomo в директории /P/y14/term3/block4/SNP/rnaseq_reads (gff и gtf формат - это почти одно и то же).
В дальнейшем я буду работать с первой репликой.
1. Анализ качества чтений.
Контроль качества чтений с помощью программы FastQC. На выходе получена html страница с отчётом. Пример выдачи этой программы вы можете увидеть в пункте 2.
Для дополнительного пояснения выдачи, см. предыдущую работу этот же пункт.
Рис. 1. Контроль качества ридов
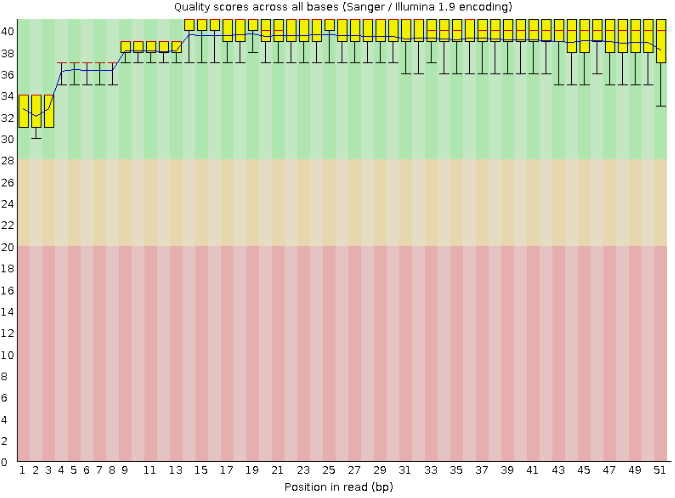
Все интерквантильные размахи качества позиций в ридах и значения качества в зелёной области (самый доверительный интервал).
Итого качество прочтения нуклеотидов во всех позициях высокое, поэтому очистка чтений, как в предыдущей работе, не потребуется.
Часть II: картирование чтений
2. Картирование чтений
Откартировать очищенные чтения с помощью программы Hisat2.
Этапы:
Сначала необходимо проиндексировать референсную последовательность; использованы те же индексные файлы, что и при картировании экзомного секвенирования (предыдущая работа).
Некоторые параметры hisat2:
-х – путь к индексу
-U – путь к чтениям
--no-softclip – запрет подрезания чтений
--no-spliced-alignment – картирование без разрывов
В этот раз не использую параметр --no-spliced-alignment, который производит картирование без разрывов. В этой работе я провожу транскриптомный анализ, а это аназин РНК,
которые могли перегруппироваться по сравнению с референсом ДНК в ходе процессинга (сплайсинг).
Затем построить выравнивание прочтений и референса в формате .sam. Запустите hisat2 с параметром --no-softclip
Сохранить вывод программы hisat2 в отдельный файл (вывод можно увидеть ниже)
|
11221 reads; of these:
11221 (100.00%) were unpaired; of these:
281 (2.50%) aligned 0 times
10795 (96.20%) aligned exactly 1 time
145 (1.29%) aligned >1 times
97.50% overall alignment rate
|
3. Анализ выравнивания
Переведите выравнивание чтений с референсом в бинарный формат .bam. Используйте пакет samtools, команда view: samtools view; Руководство по samtools.
Отсортируйте выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате в референсе начала чтения; команда samtools sort;
Проиндексируйте отсортированный .bam файл командой samtools index
Выясните, сколько чтений откартировано на геном; загляните в вывод программы Hisat2. Видим, что 281 рид не откартировался, 10795 встретились 1 раз, а 145 больше 1 раза.
В .sam файле также можно увидеть:
SRR2776256.15395984 – ID чтения
chr12 9822304 - хромосома и координата, куда «легло» чтение
100M – СIGAR: сжато кодирует информацию о выравнивании чтения
NM:i – расстояние до генома
NH:I – количество картирований для данного чтения
Часть III: Анализ bedtools
4. Подсчет чтений
Исрользуем пакет BEDTOOLS - инструмент для работы с геномными интервалами, в котором олее 35 функций + параметры
Для начала нужно перевести bam файл в формат bed. Для этого используем функцию bamtobed. В файле можем увидеть таблицу: chr, start, end, name, score, + или - цепь
Затем используем функцию intersect, задача которой наложение соответствий между двумя файлами функций. Команды можно увидеть в таблице в конце страницы
5. Анализ результатов
Таблица 1. Гены из 21 хромосомы, найденные в транскриптоме
| Ген | Координаты | Длина | Замечания | Чтений на ген |
| USP16 | 30396950-30426809 | 29859 | Этот ген кодирует дебеквивитирующий фермент, который фосфорилируется в начале митоза, а затем дефосфорилируется при метафазном / анафазном переходе. Описаны варианты альтернативного сплайсинга, кодирующие различные изоформы. Содержит 20 экзонов. + цепь | 14 |
| CCT8 | 30428126-30446118 | Длина | Этот ген кодирует тета-субъединицу 8 TCP1 шаперонина CCT, который может участвовать в транспорте и сборке вновь синтезированных белков. Альтернативное сращивание приводит к множественным вариантам транскрипции этого гена. Псевдоген, связанный с этим геном, расположен на хромосоме 1. Содержит 16 экзонов. - цепь | 188 |
Но не все чтения легли в границы генов. В конечном файле имеем chr21 30430394 30432416 sense_intronic AF129075.5, на который приходится 4 чтения, но ген CCT8 охватывает эти координаты, следовательно, можно предположить, что это
интронная область, которая была синтезирована по матрице ДНК. Возможно, что данная РНК еще не подверглась сплайсингу.
Таблица 2. Команды, выполненные в течение практикума
| Команда | Функция | Выходные файлы |
| 1. Анализ качества чтений |
| fastqc chr21.1.fastq | Вызов программы FastQC, которая контролирует качество чтений | chr21.1_fastqc.html (отчет о программе в виде html файла) |
| 2. Картирование чтений |
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 | Пакет программ, которые в данной папке, теперь доступны к вызову в putty | - |
| hisat2-build chr21.fasta chr21 | Индексирует референсную последовательность | Индексированный chr21.fasta |
| hisat2 --no-softclip -x chr21 -U chr21.1.fastq -S result.sam | Выравнивание референсной последовательности и ридов | result.sam |
| 3. Анализ выравнивания |
| samtools view result.sam -b -o result.bam | Перевод выравнивания чтений с референсом в бинарный формат .bam | result.bam |
| samtools sort result.bam -T a.txt -o sort_result.bam | Сортировка выравнивания ридов с референсом (.bam файл) по координате в референсе начала чтения | sort_result.bam |
| samtools index sort_result.bam | Индексировать отсортированный .bam файл | Индексированный sort_result.bam |
| 4. Подсчет чтений |
| export PATH=${PATH}:/P/y14/term3/block4/SNP/bedtools2/bin | Пакет программ, которые в данной папке, теперь доступны к вызову в putty | - |
| bedtools bamtobed -i sort_result.bam > result1.bed | Переводит файл формата .bam в формат .bed | result1.bed |
| bedtools intersect -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b result1.bed -u > result2.bed | Пересекает разметку генов с нашими чтениями. Параметр -u запсиывает в выходной файл только то, что хотя бы раз пересеклось | result2.bed |
Дополнительные задачи по bedtools
Таблица 3. Команды bedtools
| Команда | Задача | Дополнительные параметры | Выходные файлы |
| bedtools bamtofastq -i result.bam -fq result.fq | 1. Получите из файла с выравниванием файл с чтениями в формате fastq | -i - входной файл
-fq - выходной файл | result.fq - fastq-файл |
| bedtools getfasta -fi chr21.fasta -bed Z2.bed > Z2.fasta | 2. Получите файл с нуклеотидной последовательностью (.fasta) для одного из покрытых Вашими чтениями генов | -fi входной файл FASTA
-bed область в BED / GFF / VCF-файлах для извлечения из -fi | Z2.fasta, но во второй строчке все нуклеотиды последовательности, нет переноса |
| bedtools makewindows -g chr21.txt -w 1000000 > Z3.bed | 3. Разбейте свою хромосому на фрагменты по 1 млн нуклеотидов | -g - файл с размером генома
-w - интервал разделения | Z3.bed, где приведены интервалы длинной 1000000 |
| bedtools cluster -i result1.bed -d 100 > Z4.bed | 4. Объедините Ваши чтения в кластеры | -i - входной файл
-d максимальное расстояние между фрагментами, допускаемыми для функций объединения | Z4.bed - содержит столько же строк, сколько исходный файл, но каждому риду дан номер (кластер, к которому он принадлежит) |
| randomBed -g chr21.txt -l 200 -n 1000 > Z5.bed | 5. Наберите из Вашей хромосомы 1000 случайных фрагментов по 200 нуклеотидов | -l - длина сгенерированных интервалов
-n - число интервалов
-g - файл с размером генома | Z5.bed с нужными фрагментами |
Замечания:
2. Содержимое файла Z2.bed:
chr21 30396950 30426809 protein_coding USP16
3. Содержимое chr21.txt (разделено табулятором):
chr 46709983
Это длина хромосомы в нуклеотидах. В итоге получилось 47 интервалов.
Задания на будущее:
6. Получите координаты 3`-области одного из покрытых Вашими чтениями генов длиной в 1000 нуклеотидов.
7. Получите координаты одного из покрытых Вашими чтениями генов, расширенные на 1000 нуклеотидов в обе стороны.
8. Получите координаты одного из покрытых Вашими чтениями генов, сдвинутые на 500 нуклеотидов ближе к началу хромосомы.
9. Получите непересекающиеся фрагменты, соответствующие области, покрытой Вашими чтениями
10. Получите файл с координатами интервалов, покрытых Вашими чтениями, с информацией о покрытии в любом формате.
|



