| | | |
Комплексы ДНК-белок |
Вернуться на страницу семестра Предсказание вторичной структуры заданной тРНКПрограмма einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях. С её помощью я попробовала найти возможные комплементарные участки в последовательности исследуемой тРНК 1O0B. В таблице 1 я сравнила с их описанием, полученным ранее с помощью find_pair в предыдущей работе, и результатов, выданным программой RNAfold из пакета Viena Rna Package, которая реализует алгоритм Зукера. тРНК 1O0B в fasta-формате
Предсказание вторичной структуры тРНК путем поиска инвертированных повторов с помощью программы einverted из пакета EMBOSS (u заменены программой на t)
Рисунок 1. Предсказанная программой RNAfold вторичная структура тРНК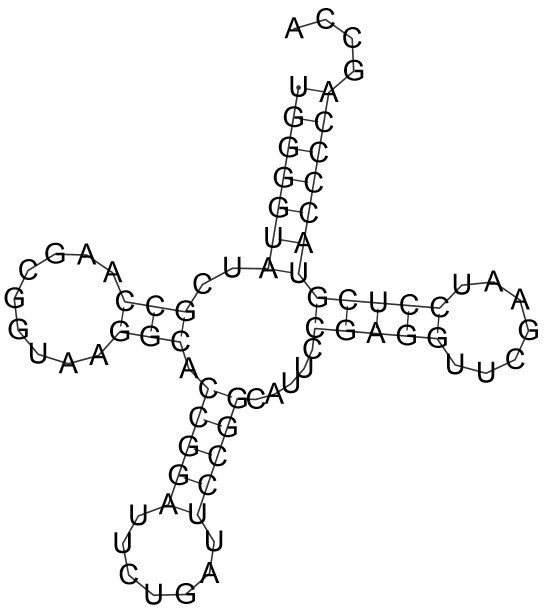
Из таблицы видно, что программа RNAfold, основанная на алгоритме Зукера, достаточно хорошо предсказывает возможные комплементарные участки молекулы тРНК. Таблица 1. Реальная и предсказанная вторичная структура тРНК из файла 1O0B.pdb
Поиск ДНК-белковых контактов в заданной структуреСсылка на скрипт В апплете Jmol приведены модели ДНК обычная, проволочная, выделены множество атомов кислорода 2'-дезоксирибозы, множество атомов кислорода в остатке фосфорной кислоты, множество атомов азота в азотистых основаниях. Контакты разного типа в комплексе 1O0B.pdbБудем считать полярными атомы кислорода и азота, а неполярными атомы углерода, фосфора и серы. Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5 (Å). Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5 (Å). Рисунок 2. Атомы принадлежащие большой и малой бороздкам
Таблица 2. Атомы азотистых оснований принадлежащие большой и малой бороздкам (рисунки взяты со страниц однокурсников)
Используя данные о бороздках и команды within, define в Jmol, составим таблицу. При этом мы видим, что для некоторых атомов и на рисунке, и в таблице точно не определена принадлежность к бороздке. Будем считать, что для пуриновых оснований она, как на рисунках, а для пиримидиновых N1, N3 принадлежат малой бороздке. Таблица 3.
Ссылка на скрипт Из таблицы можно сделать вывод, что в ДНК-белковые контактах преобладают неполярные взаимодействия, причём, если говорить о сахаро-фосфатном остове, то большая часть взаимодействий приходится на остатки фосфорной кислоты (здесь играет роль то, что они направлены наружу от ДНК и выходят из неё дальше, чем дезоксирибоза). Что касается бороздок ДНК, то значительная часть приходится на большую бороздку, что логично из геометрических и стерических рассуждений - белку легче попасть в большую бороздку, чем в маленькую. Рисунок 3. ДНК-белковые контакты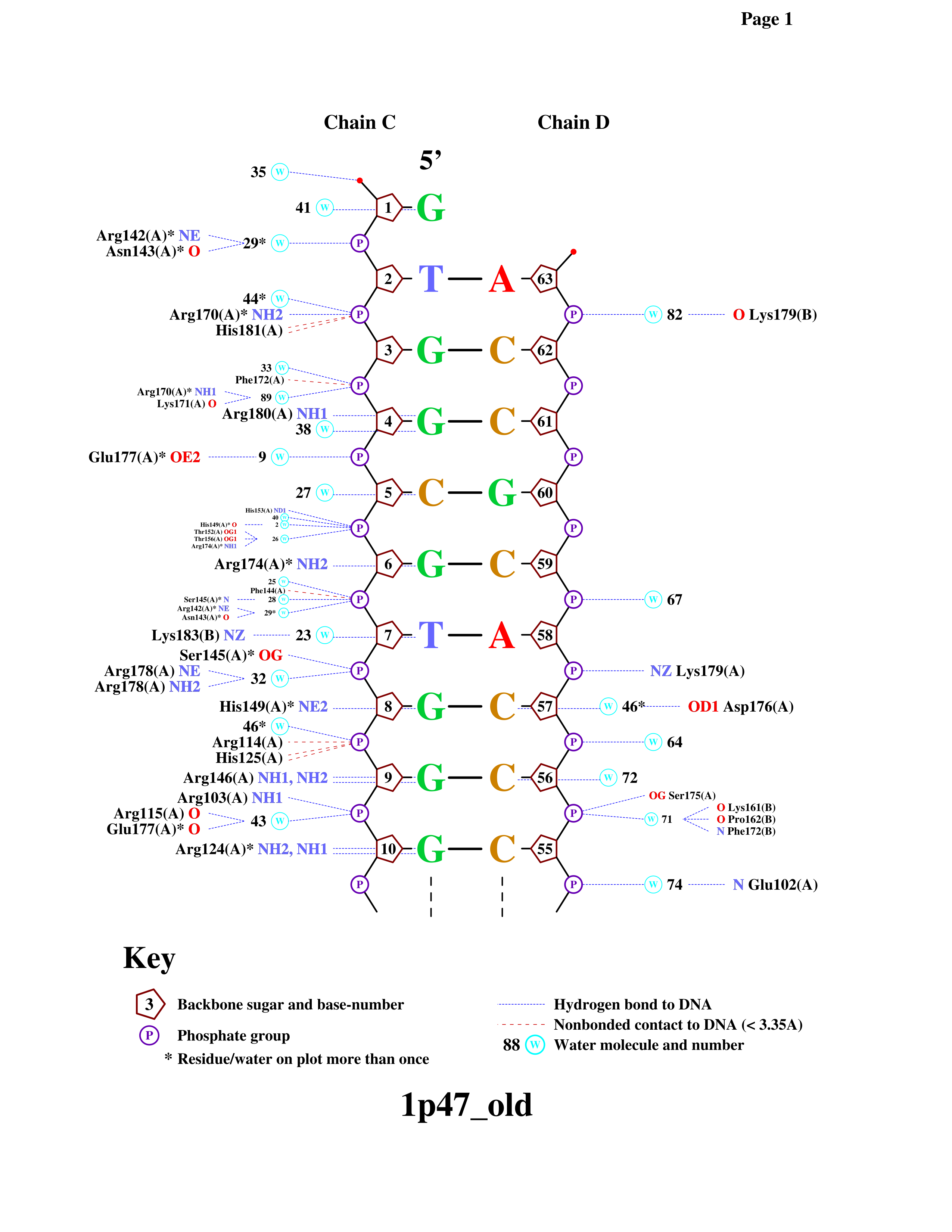
Программа nucplot, предназначенная для визуализации контактов между ДНК и белком, запускается на сервере kodomo. Программа работает только со старым форматом PDB (используйте программу remediator). Выберем аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК. Максимальное число связей для остатков на данном рисунке - 2. Выберем и рассмотрим аргинин 146, который взаимодействует с гуанином 9. При работе в Jmol действительно можно убедиться, что расстояния между N1 и N2 атомами аргинина и O6 гуанина достаточно малы, чтобы считать это полярной связью. Рисунок 4. [ARG]146:A и [DG]9:C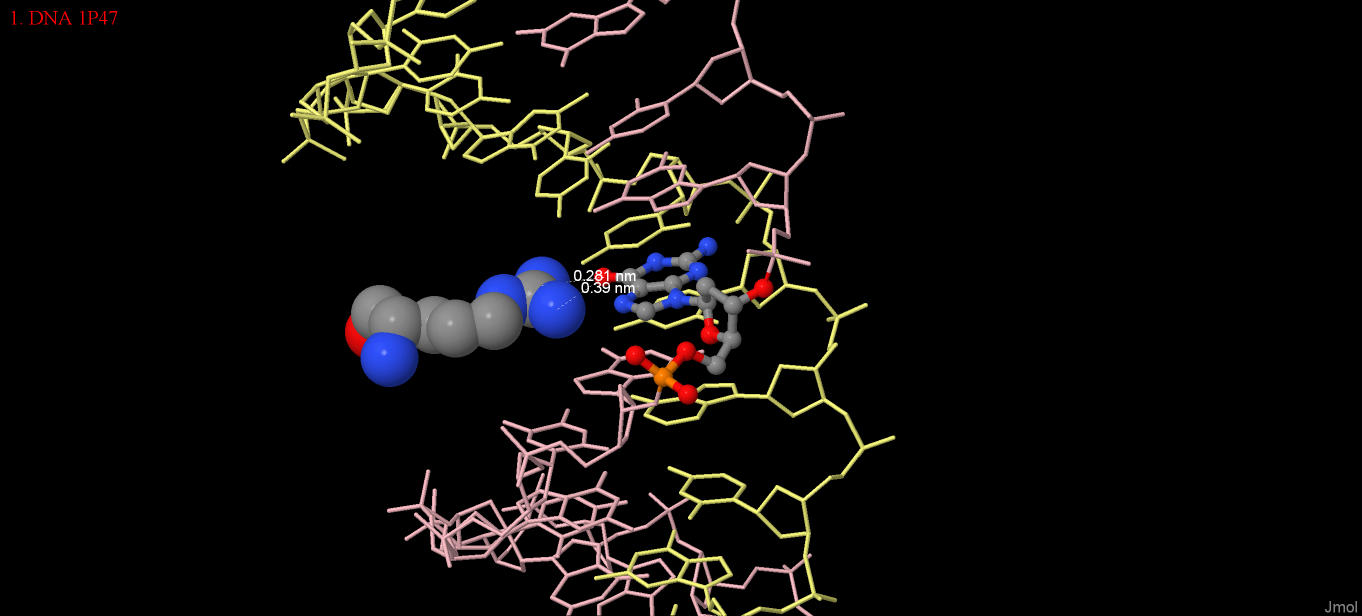
Для распознавания последовательности ДНК самыми важными мне кажутся остатки, которые связаны с нуклеотидами несколькими связями и к тому же идут друг за другом. Такие, например, гистидин 146, аргинин 146 и 124 цепи А. Они связываются соответственно с 8, 9, 10 гуанинами цепи ДНК, получается довольно специфичный способ узнавания. Рисунок 5. [ARG]124:A, [ARG]146:A, [HIS]149:A и цепь C ДНК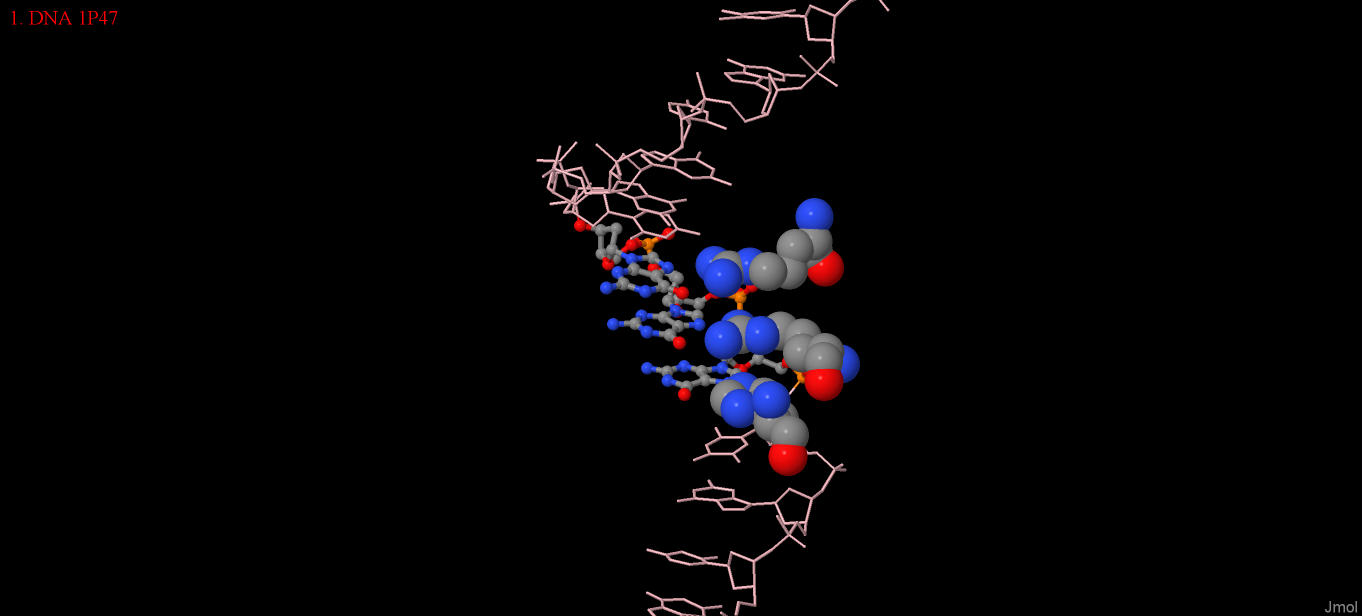
|