Семестр 3, практикум 8
Назад на учебную страницу Птицыной ЕленыНуклеотидные банки данных
В этом практикуме мы учились использовать нуклеотидные банки данных.
Задание 1.
Таксономия и функция определялись для нуклеотидной последовательности cons.fasta, полученной в практикуме 6. Для этого использовался blastn c алгоритмом поиска megablast, оптимизированным для поиска очень близких гомологов, длина слова 28, поиск по базе nt (Nucleotide collection), так как это очень обширная, объединяющая база (в другой базе, например, Refseq, нужного организма может просто не быть).
Результаты представлены на странице.
100 первых находок BLAST идентифицируют последовательность как фрагмент последовтельности, кодирующей 1 субъединицу цитохромоксидазы (CO1).
503 балла и больше набрали находки, относящиеся к роду Modiolus из брюхоногих моллюсков. Почти все они принадлежат Modiolus modiolus, некоторые - Modiolus kurilensis, 4 находки относятся к Modiolus comptus и 1 находка к Modiolus nipponicus. Нижнюю часть списка образуют виды рода Bathymodiolus, в основном Bathymodiolus thermophilus, в некоторых случах - Bathymodiolus securiformis (1 находка), Bathymodiolus septemdierum (1 находка), Bathymodiolus tangaroa (1 находка), Bathymodiolus sp. (1 находка). Между ними вклинивается Modiolus rumphii со счётом 457 баллов. Последним в списке идёт другой вид Брюхоногих моллюсков - Gigantidas crypta (427 баллов).
Далее, выбрав определенные последовательности (например, все первые 100), можно перейти на вкладку Таксономия и убедиться в том, что все 100 первых находок принадлежат семейству брюхоногих моллюсков Митилиды (Mytilidae), больше всего находок в роду Modiolus и наибольшим весом обладают выравнивания с последовательностями Modiolus modiolus, и их больше всего (рис.1).

Открыв название вида, видим страницу, содержащую полное таксономическое положение Modiolus modiolus (рис.2): cellular organisms; Eukaryota; Opisthokonta; Metazoa; Eumetazoa; Bilateria; Protostomia; Lophotrochozoa; Mollusca; Bivalvia; Pteriomorphia; Mytiloida; Mytiloidea; Mytilidae; Modiolinae; Modiolus (https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=40256).

Итак, можно сделать вывод: введенная последовательность вероятнее всего 1 субъединицу цитохромоксидазы (CO1) моллюска Modiolus modiolus.
Также есть возможность посмотреть дерево, открыв ссылку Distance tree of results (рис. 3).

При нажатии на круги клады дерева отображаются более детально:

Задание 2.
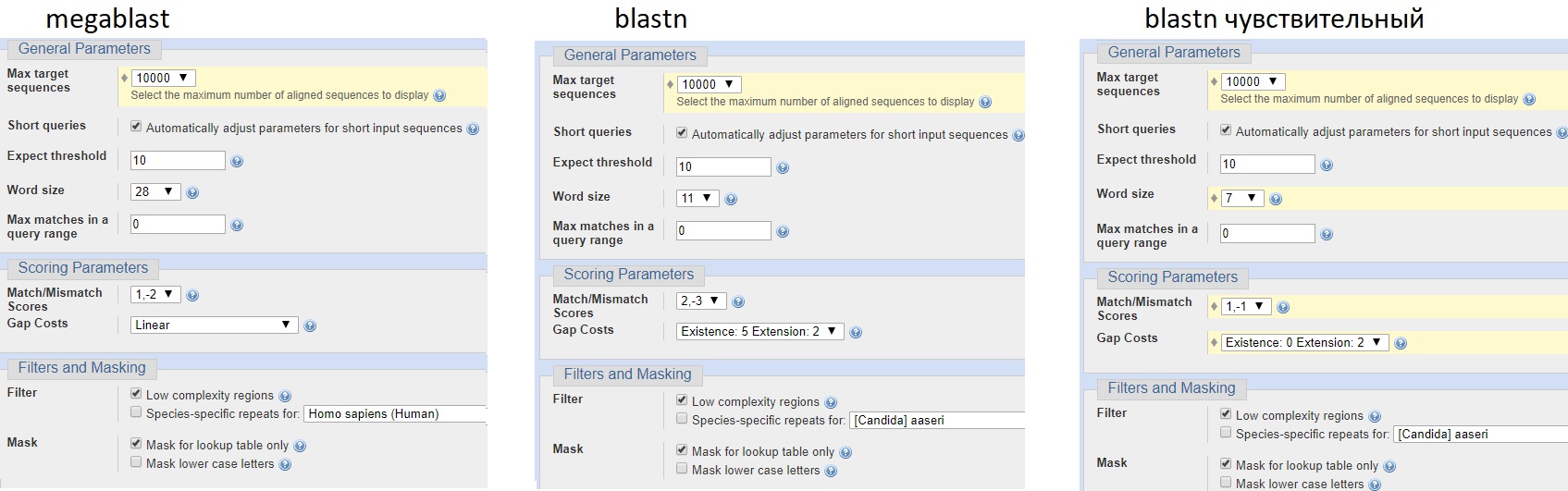
В этом задании нам надо было сравнить результаты работы BLAST для 3 алгоритмов.
Консенсунсная последовательность из практикума 6: cons.fasta
Сравнение некоторых результатов работы ( страница) показано ниже (Таблица 1).
Таблица 1. Некоторые характеристики результатов работы алгоритмов BLAST для первой последовательности.| megablast | blastn | blastn чувствительный | |
| Число находок для посл.1 | 415 | 4989 | 4996 |
| Max Score для посл.1 | 1170-58,4 | 1148-246 | 742-282 |
| Количество находок с нулевым E-value | 72 | 76 | 48 |
| E-value для одной из находок | 5e-148 | 2e-176 | 1e-129 |
| Query cover для одной из находок | 96% | 97% | 97% |
Megablast находит меньше последовательностей, чем blastn с параметрами по умолчанию и чувствительный, поскольку предназначается для поиска очень близких последовательностей, а blastn с параметрами по умолчанию находит меньше последовательностей, чем blastn чувствительный, что логично. По тем же причинам убывает верхняя граница Max Score, а нижняя поднимается. Чувствительность алгоритмов лучше всего иллюстрируется сравнением E-value для одной находки: чем меньше E-value, тем чувствительнее алгоритм (см. пример в таблице для Modiolus comptus isolate MoC-2 voucher LSGB41210-2 cytochrome c oxidase subunit I (COI) gene, partial cds; mitochondrial (GQ480313.1)).
Одна из CDS из практикума 7: cds.fasta
Параметры опять были взяты теми же. Находок было значительно меньше, они представлены на странице, обобщены в таблице 2.
Таблица 2. Некоторые характеристики результатов работы алгоритмов BLAST для второй последовательности.| megablast | blastn | blastn чувствительный | |
| Число находок | 46 | 112 | - |
| Max Score | 4488-248 | 4383-46,4 | - |
| Количество находок с нулевым E-value | 34 | 45 | - |
| E-value для одной из находок | 9e-61 | 4e-91 | - |
| Query cover для одной из находок | 76.81% | 72.28% | - |
В третьем столбце значения не приведены, так как программа говорила о превышении лимита времени анализа. Таким образом, мы убеждаемся в том, что для оптимизированного поиска в BLAST с применением высокочувствительных алгоритмов необходимо выставлять ограничения на Таксономию.
Выставив ограничения на Fuselloviridae, мы результаты (страница) , обощённые в таблице 3.
Таблица 3. Некоторые характеристики результатов работы алгоритмов BLAST для второй последовательности c ограничением на таксон.
| megablast | blastn | blastn чувствительный | |
| Число находок | 36 | 45 | 47 |
| Max Score | 4488-248 | 4383-28,3 | 2808-29,4 |
| Количество находок с нулевым E-value | 33 | 35 | 37 |
| E-value для одной из находок | 3e-66 | 1e-96 | 8e-162 |
| Query cover для одной из находок | 19% | 52% | 64.02% |
Здесь опять иллюстрируется чувствительность алгоритмов (см. пояснения к первой последовательности, правда, между blastn по умолчанию и blastn чувствительный наблюдается нарушение строгого подъёма нижней границы Max Score, но разница незначительная). Иногда одна и та же последовательность имеет не просто различающееся, а то ненулевое, то нулевое E-value (например, у Sulfolobus spindle-shaped virus Kamchatka-1, complete genome (AY423772.1) по megablast E-value ненулевое, а по другим двум алгоритмам нулевое, причём в случае blastn чувствительного она не последняя в списке находок с нулевым E-value - это очень значимое отличие, так как исследователь, визуально анализируя выдачу, в случае ненулевого E-value может просто временно отсечь в рассуждениях эту последовательность - но вдруг она важна?).
На небольшом числе находок удобно проанализировать положение отдельных находок. В верхней части списки совпадают (хотя так бывает не всегда - например, для первой последовательности порядок сверху слегка различался), а далее начинаются перестановки, появления новых находок, и т.д. (страница).
Задание 3.
Для нахождения гомологов белков из данной сборки X5.fasta мы сначала скопировали /P/y18/term3/block2/X5.fasta в свою папку, потом ввели команду makeblastdb -in X5.fasta -dbtype nucl -out sb . Далее зашли на сайт Uniprot, ввели названия 3 интересных белков, которые почти наверняка встречаются у всех эукариот ( |P62828|RAN_RAT GTP-binding nuclear protein Ran, |P50579|MAP2_HUMAN Methionine aminopeptidase 2, |Q15418|KS6A1_HUMAN Ribosomal protein S6 kinase alpha-1 ), выбрали из списков по варианту, скачали в формате fasta последовательности (ran.fasta, map.fasta (1 изоформа), kin.fasta) . Далее ввели команду tblastn -query ran.fasta -db sb -out ran.out , для двух других файлов аналогично. Получены файлы: ran.out, map.out, kin.out.
Сравнение находок представлено в Tаблице 4.
Таблица 4. Результаты поиска гомологов в данной сборке для трех белков.| Число находок | Max.score | Min.score | Min.E-value | Max.E-value | Max.identity | Min.identity | |
| ran.out | 28 | 352 (scaffold-26) | 26.9 (unplaced-979) | 2e-110 | 9.3 | 75% | 33% |
| map.out | 3 | 493 (scaffold-693) | 29.3 (scaffold-17) | 3e-155 | 6.1 | 64% | 45% |
| kin.out | 93 | 320 (scaffold-26) | 28.1 (scaffold-208) | 3e-92 | 8.4 | 52% | 40% |
GTP-binding nuclear protein Ran из крысы - ГТФаза, участвующая в нуклеоцитоплазматическом транспорте. Например, многие ядерные импортёры связывают свои субстраты только в присутствии Ran-ГТФ, а высвобождают при гидролизе ГТФ, связанного с Ran. Также изменение конформации Ran при связывании с определённым белком влияет на ядерный экспорт. 75% identity - хорошие значения, вполне подтверждающие присутствие гомолога в неаннотированном геноме.
Methionine aminopeptidase 2 из Homo sapiens - фермент, контрансляционно отщепляющий N-концевой метионин при синтезе белка. Достаточно хорошее E-value и 64% identity указывают на наличие гомолога в сборке.
Ribosomal protein S6 kinase alpha-1 из человека - рибосомальная протеинкиназа, участвующая в сигнальном пути ERK. Из-за весьма низкого значения identity гомология ставится под вопрос. Вероятно, ввиду большой распрострённости и востребованности киназы очень разнообразны.
Задание 4.
В этом задании нам надо было сначала взять из какой-либо сборки контиг или скэффолд длины порядка десятков тысяч пар нуклеотидов, в котором ещё не аннотированы гены белков, а потом найти правдоподобный ген. Для этого мы использовали сборку X5 Amoeboaphelidium protococcarum с kodomo, с помощью команды infoseq X5.fasta -only -name -length выяснили длины фрагментов, выбрали один (scaffold-698 длиной 23460) и вставили его в blastx (переводит изучаемую нуклеотидную последовательность в кодируемые аминокислоты, а затем сравнивает её с имеющейся базой данных аминокислотных последовательностей белков), выбрали поиск по RefSeq и поставили ограничение на Опистоконт. Результаты представлены на странице. Все находки имели нулевое E-value и большинство указывали на то, что в данном скэффолде содержится ген субъединицы гамма 2 фактора элонгации транскрипции. Странно, что третья находка очень выпадающая - P-loop containing nucleoside triphosphate hydrolase protein. По данным UNIPROT, он ещё не аннотирован и связывает актиновые филаменты. Возможно, в будущем кто-нибудь выявит связь между ним и фактором элонгации транскрипции - скорее, сходство с P-loop containing nucleoside triphosphate hydrolase protein является результатом коэволюции.