Практикум 9. Белковые домены

С помощью Pfam мы выбрали доменную архитектуру, содержащую домен пектинэстеразы: Pectinesterase (PF01095) и Lipase_GDSL_2 (PF13472). Pfam показывает, что 49 последовательностей имеют такую структуру:


Далее мы искали белки бактерий, содержащие эти два домена, в Uniprot:
database:(type:pfam pf01095) database:(type:pfam pf13472) taxonomy:"Bacteria [2]"
Uniprot нашел 386 последовательностей. Они были скачаны в виде файла Excel. Потом мы посмотрели, сколько белков бактерии находит Uniprot для каждого домена по отдельности. Для PF01095 - 7201, для PF13472 - 105074.
В файле Excel (лист "Data") построили условную "функцию распределения" (в абсолютных количествах) для длин белков и по ней оценили типичную длину: 80% белков имеют длину 520-640 а.к.о. При просмотре информации на Pfam оказалось, что есть еще одна доменная архитектура с такими же двумя доменами, но в обратном порядке! Ей отвечает несколько меньше белков (по Pfam их 14), чем выбранной нами.
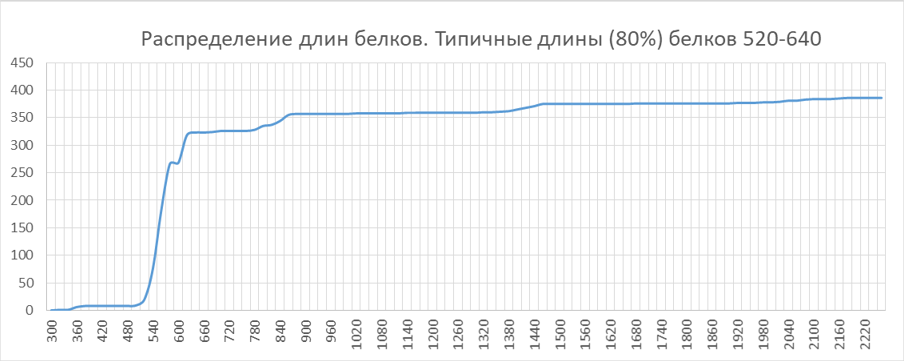

Вот еще гистограмма распределения длин белков. Она соответствует "плотности" указанной выше "функции распределения".
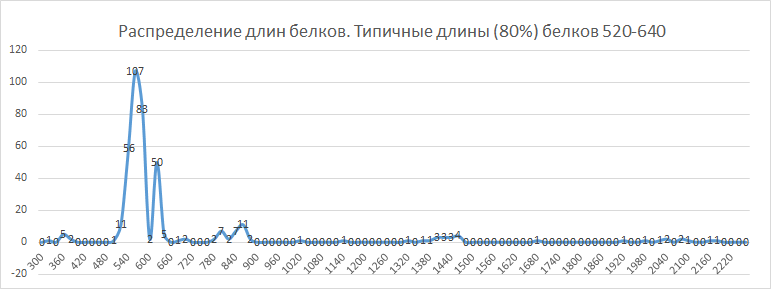
Мы выбрали (лист "Sample") 55 белков, принадлежащих разным бактериальным отделам и отметили их на листе "Data".
Далее нам было интересно найти белки с нашей доменной архитектурой среди белков, содержащих один из доменов. Сначала мы скачали с сайта Uniprot последовательности белков, имеющие в составе домен PF01095 и поместили их в файл SEQ.fasta. Затем мы пользовались пакетом программ HMMER для создания профиля и подсчета веса выравнивания последовательности с профилем. Для построения профиля мы предварительно получили из Uniprot последовательности для нашей выборки белков двухдоменной структуры и выравняли их в программе Jalview. Удалили N- и C-концы, чтобы по краям остались консервативные блоки; удалили последовательности, не имеющие какого-нибудь из краевых консервативных блоков. Сохранили выравнивание в файл PE_sample_pr9.fa. Далее построили на основе этого выравнивания профиль (с функцией для глобального выравнивания):
hmm2build -g profile.hmm PE_sample_pr9.fa
Далее откалибровали профиль на основе последовательностей в случайном банке данных (нормализация рассчета веса и evalue):
hmm2calibrate profile.hmm
Наконец, выравняли по очереди все найденные последовательности белков (с одним доменом) с профилем и высчитали вес выравниваний и evalue с учетом калибровки:
hmm2search profile.hmm SEQ.fasta > info.txt
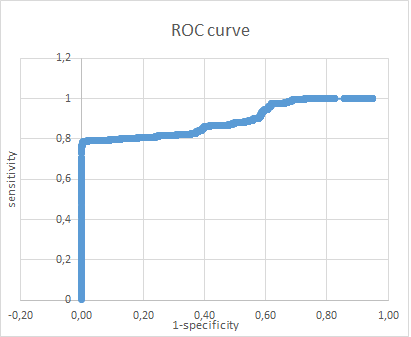
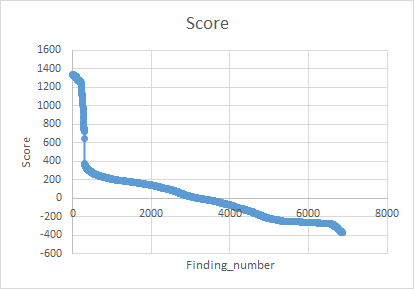
Информация (info.txt - этот файл не вижу смысла вставлять, там есть нужная нам информация о выравниваниях и собственно сами выравнивания) выдалась о 6865 последовательностях из 7201. Поэтому при учете истинно негативных ответов (без нашей доменной архитектуры) мы учитывали 346 последовательностей, не прошедших evalue (10 по умолчанию) и очевидно негативных. Далее для каждой строчки (где были написаны вес и evalue для выравнивания каждой последовательности) считали, сколько было бы истинно позитивных и истинно негативных результатов, если в качестве теста на доменную архитектуру мы брали бы прохождение порога веса, указаного в данной строке. На основе этого считали для каждой строки чувствительность и специфичность. Потом построили график ROC-кривой и вычислили площадь под ней. Она оказалась равна 0.90, что достаточно неплохо. В качестве порога выбрали вес, соответствующий строке, сумма чувствительности и специфичности в которой максимальны. Пороговй вес получился равен 351.5, evalue - 1е-102. Все рассчеты производились в Excel (см. файл ниже). График распределения весов для находок находится в этом же файле.
| # | Test+ | Test- | sum |
|---|---|---|---|
| Positive | 302 | 84 | 386 |
| Negative | 9 | 6806 | 6815 |
| sum | 311 | 6890 | 7201 |
Эта таблица (а еще представленные в файле выше и в тексте выше значения чувствительности и специфичности) говорят о том, что у нашего теста оказалась весьма хорошая специфичность, а чувствительность похуже. Я считаю, что специфичность в данной задаче нужнее чувствительности: если, например, у нас стоит задача множественного выравнивания, то многие последовательности без нужной архитектуры внесут серьезный шум и не позволят выйти адекватному выравниванию, да и воспринять выравнивание, в которм 90% последовательностей ложноположительны, можно превратно: посчитать, что меньшинство истинно положительных ложны (такое МОЖЕТ быть, если в выравнивание попало много белков с другой, более распространенной доменной архитектурой).