Методы и их применения в Excel
Ссылка на изначальные таблицы с 4 плазмидами и хромосомой: {ссылка на xlsx-файл}.
Ссылка на таблицу с выполненными заданиями: {ссылка на xlsx-файл}.
Проверка гипотезы о том, что гены распределены между двумя цепями ДНК случайно с вероятностью 0.5.
Я использовал встроенную функцию 'БИНОМ.РАСП'. Проанализировав в таблице данные, однозначно утверждать правдоподобность или
неправдоподобность гипотезы нельзя, так как отсутствует выборка, по которой необходимо анализировать гипотезу. Однако, можно
предположить, что при возрастании количества генов на прямой и обратных цепях, пик, получающийся на графике биномиального распределения становится резче,
следовательно всё больше значений отдалённых от элемента, занимающего среднее положение в цепи, будут иметь значения, стремящиеся к нулю.
Тем не менее, при попадании значений под пик графика, они мгновенно стремятся к максимальному значению. Также, отмечу, что прослеживается закономерность: чем больше разница между значениями двух
генов, тем меньше процент p-value. Взглянем на некоторые графики.
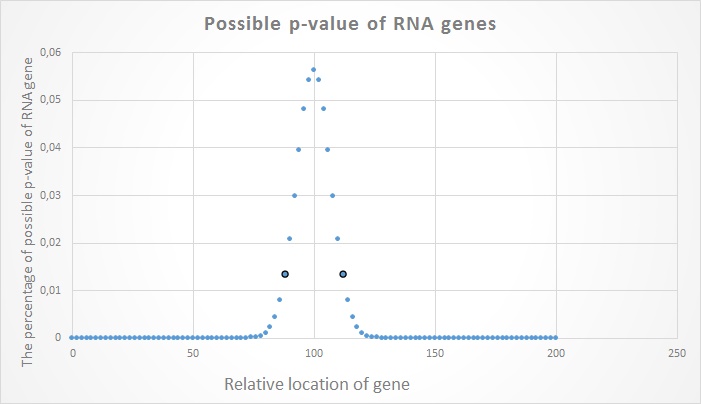
Как видно из Рис.1 значения, отмеченные чёрными точками, расположены достаточно далеко от 0, но не так
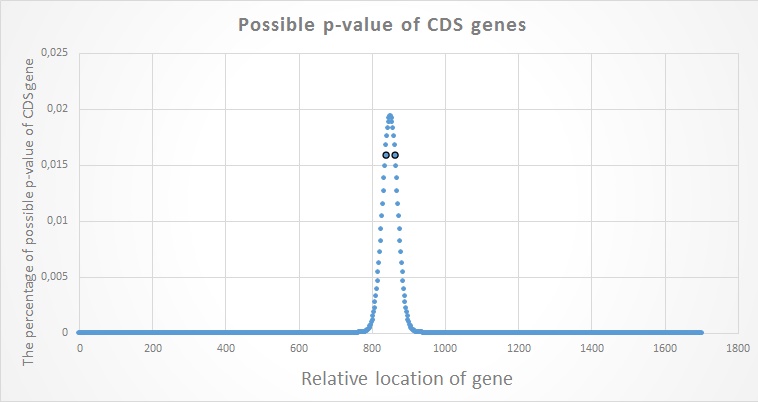
близко к пику, как хотелось бы. Выборка по RNA состоит из 200 генов, то есть разность между генами составляет довольно большой процент относительно всего количества генов. Взглянем на Рис.2. Ситуация получше, несмотря на выборку, уже состоящую
из 1703 генов(!). Значения, отмеченные чёрными точками имеют почти максимальный процент возможного отхождения от элемента, занимающего
среднее положение в цепи. Также, видно, что огромное количество значений составляет линию тренда у 0, что подтверждает написанное выше.
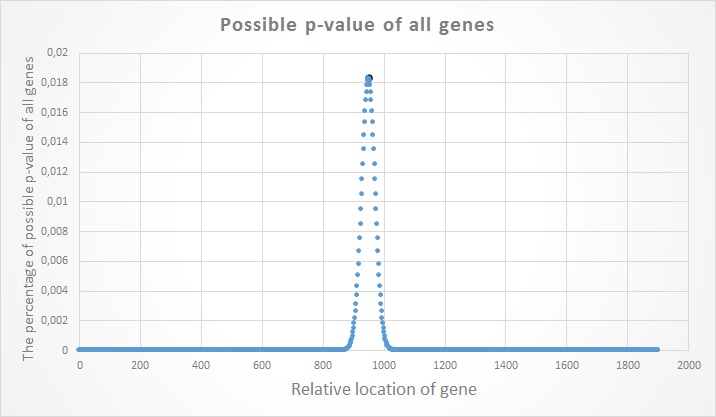
И наконец рассмотрим псоледний график, построенный по всем генам, вне зависимости от типа гена.
На Рис.3 мы видим 'идеальное' распределение, чёрные точки (их там 2) являются максимальными значениями,
которые могут быть (*на самом деле возможен ещё более 'идеальный' вариант распределения, например одна точка, являющаяся максимальным занчением,
однако при выборе шага, было взято значение 2, а не 1, поэтому 'идеальное' одиночное значение не показывается при таком выборе шага).
Подведём итог: биномиальное распределение для RNA-точек (112 и 88) равно 10.364%, для CDS (838 и 864) - 54.454%, для ALL (950 и 952) - 98.171%.
*Если остались какие-то вопросы, лучше заглянуть в таблицу.
Подсчёт количества квазиоперонов и пересечений генов в геноме Leuconostoc citreum KM20.
Схема подсчёта, использованные формулы и методы приведены в таблице.
Некоторые пояснения: я считал квазиопероны как по плазмидам, так и по хромосоме отдельно, по цепям также делал разделение, позже суммировал полученные результаты.
Интересно, что процент содержания квазиоперонов в геноме равен 21.923%, что составляет почти четверть всего генома бактерии. Это неудивительно,
ведь согласно заданию, расстояние между квазиоперонами не должно превышать 100 нуклеотидов, а каждый ген входит в какой-либо квазиоперон.
Изменяя значение расстояния со 100 на 50 и 200, получаем 29.231% и 14.945% соответственно. Хочется подставить значение равное 20, получим, что при данном значении процентное содержание квазиоперонов в геноме равно 34.945%.(*Видно, что процентное содержание квазиоперонов растёт не так быстро, как ожидалось) Так происходит в связи с тем, что уменьшая расстояние, мы получаем большее количество 'одиночных' квазиоперонов, и наоборот.
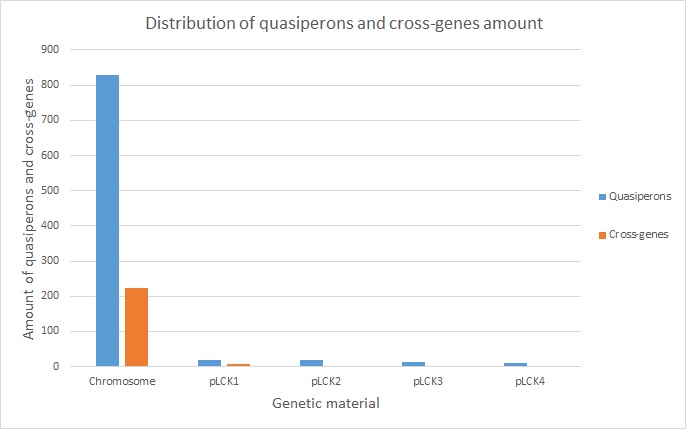
Схема нахождения и расчёты приведены в таблице. Некоторые комментарии: как видно из Рис.4, самое большое количество пересечений генов наблюдается в хромосоме (224), в остальном генетическом материале вместе взятом, 4 плазмиды - (13). Процент пересечений от всех генов составляет 12.461%. Объяснение случаев, когда длина кодирующей последовательности не кратна 3. В таблице указана схема проверки кратности 3 всего генетического материала. Делать однозначные выводы сложно, однако данные получились говорящими сами за себя. Только RNA-гены не кратны 3, причём превалируют гены НЕ кратные, нежели кратные в RNA-генах. Единственное логическое объяснение, которое сразу приходит на ум, - это интроны [1]. Действительно, интроны могут присутствовать в RNA-генах у прокариот. Случаев же, когда интроны присутствуют в CDS-генах куда меньше, однако тоже таковые имеются. Анализируемая мною бактерия идеально подходит под теоретическую основу. Так что скорее всего именно интроны являются 'виновниками' некратности 3 кодирующей последовательности. (*Чтобы сделать адекватный вывод, конечно, необходима выборка для прослеживания закономерности)
Источники информации:
[1]: Wikipedia (Intron)
[2]: NCBI (Data)
Ссылка на таблицу с выполненными заданиями: {ссылка на xlsx-файл}.
Проверка гипотезы о том, что гены распределены между двумя цепями ДНК случайно с вероятностью 0.5.
 |
 |
 |
 |
Изменяя значение расстояния со 100 на 50 и 200, получаем 29.231% и 14.945% соответственно. Хочется подставить значение равное 20, получим, что при данном значении процентное содержание квазиоперонов в геноме равно 34.945%.(*Видно, что процентное содержание квазиоперонов растёт не так быстро, как ожидалось) Так происходит в связи с тем, что уменьшая расстояние, мы получаем большее количество 'одиночных' квазиоперонов, и наоборот.
Схема нахождения и расчёты приведены в таблице. Некоторые комментарии: как видно из Рис.4, самое большое количество пересечений генов наблюдается в хромосоме (224), в остальном генетическом материале вместе взятом, 4 плазмиды - (13). Процент пересечений от всех генов составляет 12.461%. Объяснение случаев, когда длина кодирующей последовательности не кратна 3. В таблице указана схема проверки кратности 3 всего генетического материала. Делать однозначные выводы сложно, однако данные получились говорящими сами за себя. Только RNA-гены не кратны 3, причём превалируют гены НЕ кратные, нежели кратные в RNA-генах. Единственное логическое объяснение, которое сразу приходит на ум, - это интроны [1]. Действительно, интроны могут присутствовать в RNA-генах у прокариот. Случаев же, когда интроны присутствуют в CDS-генах куда меньше, однако тоже таковые имеются. Анализируемая мною бактерия идеально подходит под теоретическую основу. Так что скорее всего именно интроны являются 'виновниками' некратности 3 кодирующей последовательности. (*Чтобы сделать адекватный вывод, конечно, необходима выборка для прослеживания закономерности)
Источники информации:
[1]: Wikipedia (Intron)
[2]: NCBI (Data)
⌘
© Emir Radkevich, 2016