Нуклеотидный blast
Задание 1
В Практикуме 6 после редактирования результатов прочтения хроматограммы секвенирования была получена последовательность ws-3016f.fasta.
По ней был запущен алгоритм blastn (somewhat similar sequences) со следующими параметрами:
- База данных: Nucleotide collection (nr/nt)
- Максимально возможное количество находок: 20 000
Параметры алгоритма были оставлены без изменений. Значения по умолчанию:
- Порог ожидания: 10
- Длина слова: 11
- Балл за совпадение: 2
- Штраф за несовпадение: -3
- Штраф за открытие гэпа: -5
- Штраф за расширение гэпа -2
На Рис.1 можно видеть наилучшие находки blastn.
Рис. 1
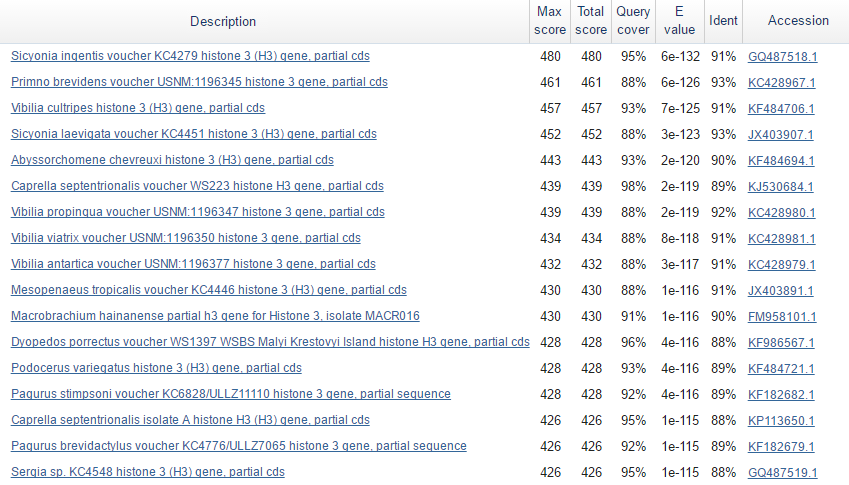
Всего было 20000 находок, что соответствует максимальному числу, которое можно было выставить в параметрах. E-value лучшей находки составил 6е-132, а худшей — 1e-65 (но тут, впрочем, query cover составлет 76%, что в целом неплохо). Все находки являются нуклеотидными последовательностями эукариот. Все они относятся к белку гистону H3.
Из всего сказанного выше можно заключить, что исследуемая последовательность также содержит часть гена, кодирующего белок гистона H3. Большое число находок связано с консервативностью аминокислотной последовательности этого белка в различных группах эукариот.
Для пяти лучших найденных последовательностей было построено выравнивание с исходной (она первая в выравнивании) в jalview (проект [jvp] доступен по ссылке). Результат можно вижеть на Рис.2.
Рис. 2

На Рис.3 изображено таксономическое положение видов из лушчих находок. В рамку обведены виды. Числа сбоку — это количество замен на 100 нуклеотидов.
Рис.3 |
Из таблицы видно, что рассмотренные лучшие находки относятся к подклассу Eumalacostraca. Отнесение же к надпорядку уже сопряжено с проблемами. У представителей надпорядка Eucarida в целом находки лучше, ибо произошло меньше замен. Но одна находка из надпорядка Amphipoda имеет 7 замен на 100 нуклеотидов, в то время как у Sicyonia laevigata (из Eucarida) этот результат хуже — 7.5. В итоге можно сказать, что уж точно последовательность получена из организма, относящегося к подтипу Ракообразных (Crustacea), классу Высшие раки (Malacoostraca), подклассу Эумалакостраки (Eumalacostraca). Думаю, впрочем, что если учитывать тот факт, что среди 5 лучших находок 2 относились к одному и тому же роду, а именно — Sicyonia, то можно с некоторой долей уверенности говорить о том, что исследуемая последовательность также принадлежит представителю этого рода. Вообще Sicyonia — это такой род креветок, так называемых ridgeback shrimps. S. ingentis показана на рисунке ниже.  |
Итоговая предполагаемая таксономия (до рода): Eukaryota; Opisthokonta; Metazoa; Eumetazoa; Bilateria; Protostomia; Ecdysozoa; Panarthropoda; Arthropoda; Mandibulata; Pancrustacea; Crustacea; Malacostraca; Eumalacostraca; Eucarida; Decapoda; Dendrobranchiata; Penaeoidea; Sicyoniidae; Sicyonia
Задание 2
Было проведено сравнение трех алгоритмов — blastn (somewhat similar sequences), discontigious megablast (more dissimilar sequences), megablast (highly similar sequences).
Таблица 1. Параметры запуска blast
| Алгоритм поиска | Database | Max target sequences | Expect treshold | Word size | Max matches in a query range | Match/Mismatch scores | Gap costs |
| blastn | Nucleotide collection (nr/nt) | 100 | 10 | 11 | 0 | 2;-3 | 5;2 |
| discontigious megablast | Nucleotide collection (nr/nt) | 100 | 10 | 11 | 0 | 2;-3 | 5;2 |
| megablast | Nucleotide collection (nr/nt) | 100 | 10 | 28 | 0 | 1;-2 | linear |
С приведёнными выше параметрами была запущена работа трёх алгоритмов, при это область поиска была ограничена таксоном Decapoda (отряд Десятиногие).
Таблица 2. Число находок
| Алгоритм | Число находок |
| blastn | 45 |
| discontigious megablast | 30 |
| megablast | 25 |
Находки по трем алгоритмам записаны в файле [xlsx] и доступны для скачивания.
Среди прочих есть находки, которые были найдены только при помощи одного алгоритма (blastn), при помощи только двух (blastn и discontigious megablast). Находки, содержащиеся в выдаче megablast, были найдены в каждом из трёх случаев.
Лучшие находки (11 первых) совпадают во всех алгоритмах. E-value у худших находок очень различается: у megablast он очень низкий (2.33е-90), у discontigious megablast тоже относительно неплохой (1.61е-77), а вот у blastn он составляет аж 6.9. Таким образом, в числе находок blastn присутствуют такие, которым лучше не доверять. К тому же, у blastn и discontigious blast минимальный query cover ~80.6%, в то время как у megablast он выше и составляет ~86%.
Итак, можно прийти к выводу о том, что megablast отсекает находки с плохим покрытием или высоким E-value и выдает только самые близкие к нашей последовательности. Это связано, помимо прочего, с высокой длиной начального слова, которая составляет 28.
discontigious megablast дает меньше находок, чем blastn, но, опять-таки, он отсек только те, что обладали не очень хорошим качеством.
Пример находки, которая была обнаружена только blastn и discontigious blast: Gennadas valens voucher HBG1132A histone 3 (H3) gene, partial cds (KP076142.1) — Max score: 383, Total score: 383, Query cover: 88%, E-value: 8e-107, Ident: 88%.
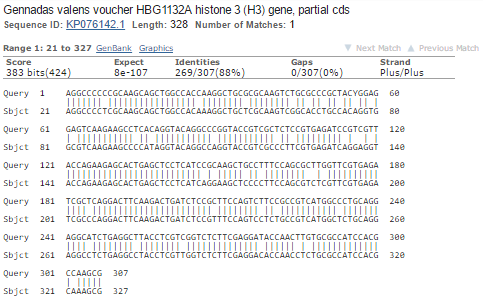
Посмотрим, почему она не попала в megablast. Из Рис.4 видно, что в ее выравнивании относительно заданной последовательности не было отрезка длиной не менее 28 нуклеотидов.
Рис. 4. Выравнивание с последовательностью, не найденной megablast
Выводы. Проведенное сравнение позволяет сделать следующие выводы о специфике работы алгоритмов поиска:
- megablast находит самые достоверные и наиболее близкие гомологи
- discontigious megablast хорошо работает в плане поиска гомологов, которые прежде уже дивергировали, и имеют как схожие, так и различающиеся участки
- blastn выдает наиболее широкий спектр последовательностей, которые могут, вообще-то, и не быть гомологами исходной или являться очень далекими гомологами
Задание 3
Мною были выбраны белки HSP7C_HUMAN, TERT_HUMAN, CISY_HUMAN. Их последовательности я скачала и объединила в единый файл формата [fasta]. Таоке объединение было проведено, поскольку это удобно, а также потому что тогда получение результата работы алгоритма blast занимает меньше времени.
Я создала локальную базу данных на основе генома Amoeboaphelidium protococarum, записанного в файле X5.fasta, следующим образом:
makeblastdb -in X5.fasta -dbtype nucl
tblasn — это программа, которая ищет по нуклеотидному банку последовательностей те, что кодируют белки, подающиеся на вход. Ей я и воспользовалась:
tblastn -query inprot.fasta -db X5.fasta -out blast.out -outfmt 7
Выдача алгоритма для трёх выбранных белков приведена на рисунках ниже.
| Белок | Выдача tblastn |
| HSP7C_HUMAN | 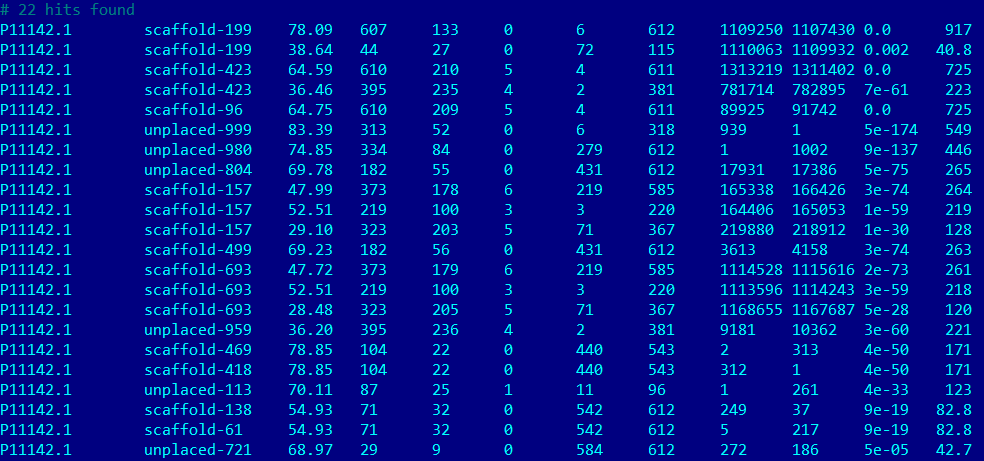 |
| Белок HSP7C[1] — это белок теплового шока. Репрессор активации транскрипции. Шаперон. Является компонентом PRP19-CDC5L комплекса, который формирует внутренню часть сплайсосомы. Этот белок необходим для активации сплайсинга пре-мРНК.
Может выполнять структурную функцию в сборке сплайсосомы, поскольку контактирует со всем остальными ее компонентами. Связывает бактериальные липополисахариды и опосредует ЛПС-идуцированную воспалительную реакцию.
В результате работы tblastn мы получили 22 находки. Первая из них, scaffold-199, представляется достаточно качественной. E-value 0.0, процент покрытия 78.09%. Последний показатель может показаться не очень высоким, но посмотрев отдельно на выравнивание (для этого scaffold-199 нужно было извлечь из файла генома), можно понять, что несовпадения сконцентрированы в конце выравнивания, поэтому, как минимум, здесь можно говорить о гомологии доменов с сохранением функции. | |
| TERT_HUMAN |  |
| Белок TERT_HUMAN[2] — это обратная транскриптаза теломеразы. Теломераза — рибонуклеопротеиновый фермент, необходимый для репликации концов хромосомы у большинства эукариот. Активен в прогениторных и раковых клетках, в отличие от обычных соматических, где практически не действует. Является
участником процесса элонгации теломер, при этом действуя как обратная транскриптаза: добавляет простые повторные последовательности к концам хромосомы, копируя образец с РНК-компонента фермента. (Катализирует РНК-зависимое удлинение 3'-конца хромосомы с помощью 6-нуклеотидной последовательности 5'-TTAGGG-3'). Каталитический цикл включает связывание праймера, удлинение праймера и высвобождение праймера по достижению конца РНК-затравки или же перенос возникающего продукта с его последующим удлинением. Играет важную роль в процессах старения и предотвращения апоптоза.
На выдаче имеем 3 находки. Лучшая из них — scaffold-17 с E-value 8e-23 и Query cover 26.58%. Посмотрим на выравнивание. Раскраска BLOSUM62 By Conservation.  Выравнивание, конечно, не самого высокого качества, но всё же встречаются участки (100-134, 381-402 и другие), на которых прослеживается сохранение мотивов. В этом случае, думаю, ответ на вопрос о наличии гомологов будет условно положительный. Вполне вероятно, что отдельные домены белка, закодированного в каком-то из генов scaffold-17, сохранили схожую с TERT_HUMAN функцию. | |
| CISY_HUMAN[3] | 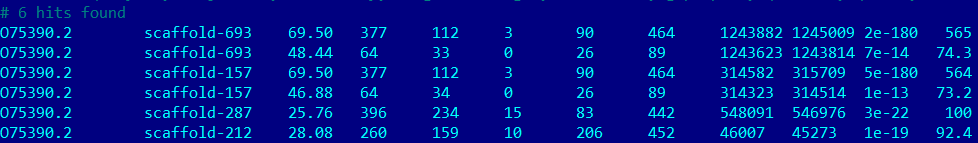 |
Белок CISY_HUMAN — это белок митохондриальной цитрат-синтазы. Он катализирует следующую реакцию:
Ацетил-КоА + H2O + оксалоацетат —> цитрат + КоАИтак, он принимает участие в метаболизме углеводов, а именно — в цикле трикарбоновых кислот на стадии получения изоцитрата из оксалоацетата. tblastn выдал 6 находок. Лучшая из них — scaffold-693 с E-value 2e-180 и Query cover 69.5%. Интересно отметить, что с ней по всем параметрам очень схожа третья по счёту находка scaffold-157, только у нее BitScore на единицу меньше, а E-value чуть выше и составляет 5e-180. Также внутри обоих скэффолдов было по две находки (соответственно, 2-ая и 4-ая). Находки из двух разных скэффолдов имели одинаковые по сути выравнивания с последовательностью CISY_HUMAN, а различались рамкой считывания при формальной трансляции нуклеотидной последовательности скэффолда в последовательность аминокислот. Выравнивания хорошего качества, поэтому, как мне кажется, здесь можно вполне утверждать наличие гомолога. Для отдельно взятого скэффолда (693-ий или 157-ой) первое выравнивание соответствует одной части белка, гомологичного CISY_HUMAN, а второе — другой. Причём эти части в последовательности CISY идут подряд, а в исследуемом геноме разнесены в разные области скэффолда. | |
Задание 4
Для выполнения задания я выбрала из генома Amoeboaphelidium protococarum скэффолд scaffold-693. Предварительно была получена информация о длинах контигов:
infoseq X5.fasta -only -name -length
А затем извлечена последовательность подходящего по длине scaffold-693:
seqret X5.fasta:scaffold-693 -out 693.fasta
По этой последовательности был запущен алгоритм megablast с ограничением на таксон Amoeboaphelidium protococarum. Результаты можно видеть на Рис. 5
Рис.5

Наблюдаем две достаточно хорошие находки (для их быстрого обнаружения и был использован megablast). Первая находка включает в себя один совпадающий участок, а вторая — два, причём первый участок из них выровнен ровно так же, как и в первой находке.
Рис. 6. Выравнивания находок из выдачи blast
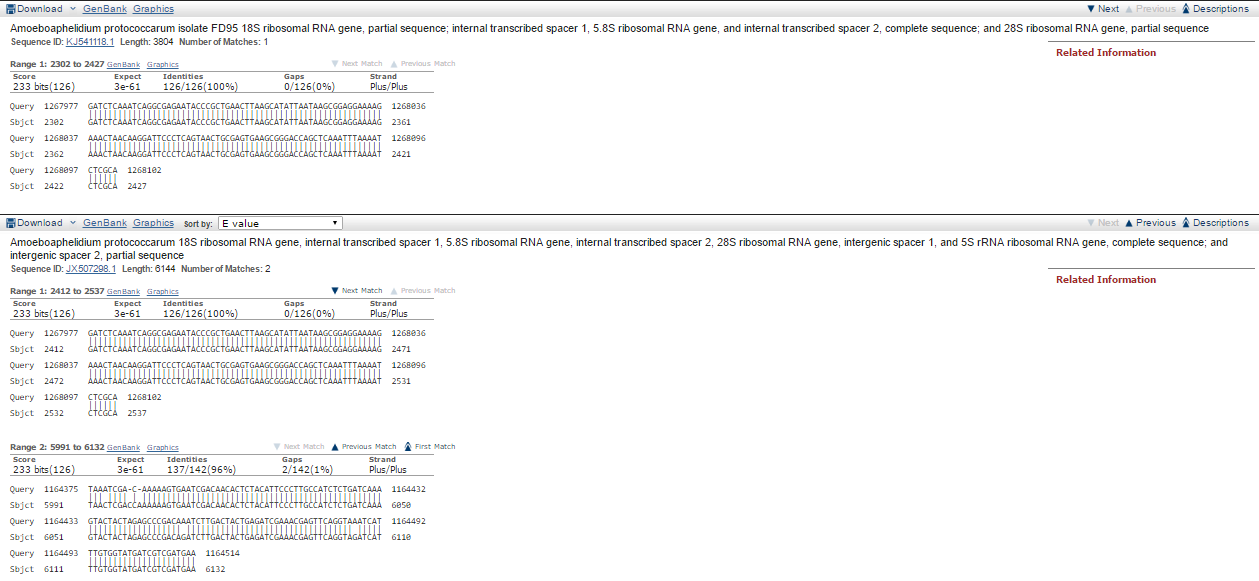
По координатам участков и аннотации последовательностей в находках можно понять, что за гены содержатся в scaffold-693. Один из найденых участков участвует в кодировании 28S-рРНК, а второй — является частью спейсерного участка[4], функция которого предположительно заключается в обеспечении выского уровня транскрипции в связанных генах.
[1] — Uniprot - P11142
[2] — Uniprot - O14746
[3] — Uniprot - O75390
[4] — Wiki