Вспомним, как с помощью команды define Jmol задавать множества атомов, а именно атомы кислорода 2'-дезоксирибозы, атомы кислорода в остатке фосфорной кислоты, атомы азота в азотистых основаниях. Создадим скрипт-файл с определениями этих множеств:script1.txt. Далее создадим скрипт, вызов которого даст последовательное изображение всей структуры, только ДНК в проволочной модели и ДНК в проволочной модели с выделенными множествами атомов, перечисленных ранее:script2.txt.
Опишем ДНК-белковые контакты в структуре 1P47. Сравним количество контактов разной природы. Будем считать полярными атомы кислорода и азота, а неполярными атомы углерода, фосфора и серы.Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5Å. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5Å.
Напишем скрипт, позволяющий определить число контактов в структуре 1P47:script3.txt. Результаты работы скрипта представлены в табл.1.
Табл.1.Контакты разного типа в комплексе .
| Контакты атомов белка | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 0 | 32 | 8 |
| остатками фосфорной кислоты | 8 | 12 | 20 |
| остатками азотистых оснований со стороны большой бороздки | 0 | 32 | 32 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 3 | 3 |
Мы видим, что полярных контактов намного меньше, чем неполярных. Чаще всего белок связывается с остатками фосфорной кислоты и остатками азотистых оснований со стороны большой бороздки. Это связано с тем, что они более доступны для контакта с белком в силу их простанственного расположения.
Используем программу nucplot для визуализации контактов между ДНК и белком в структуре 1Р47:
remediator --old "1P47.pdb" > "1P47_old.pdb" nucplot 1P47_old.pdb
Таким образом, получили схему контактов белка с ДНК.
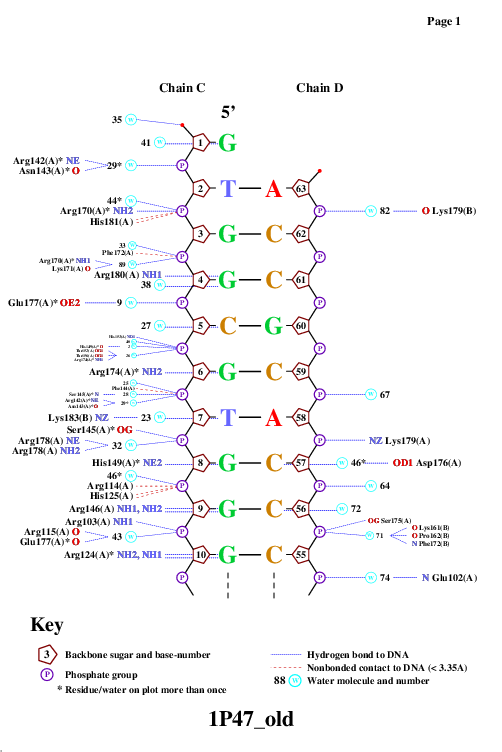
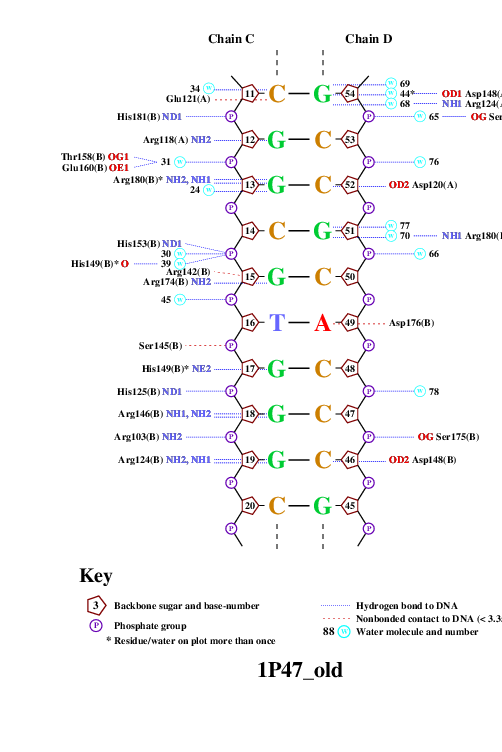
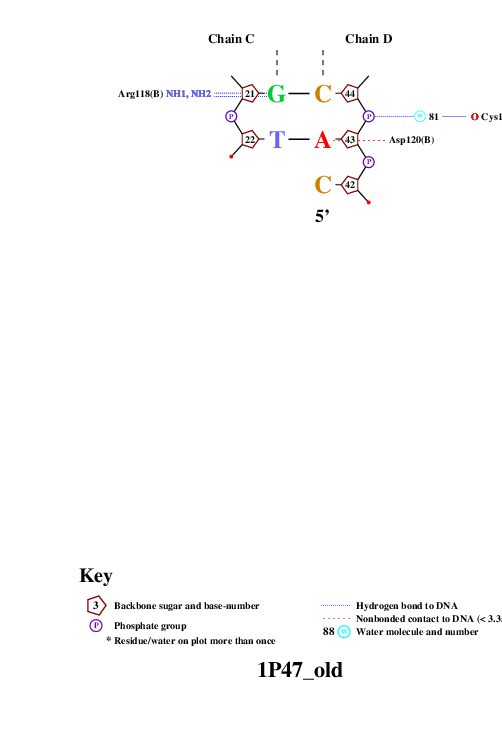
Белок, входящий в заданный мне комплекс состоит из двух одинаковых цепей А и В. На схеме можно заметить, что аминокислотные остатки с наибольшим числом контактов с ДНК - Arg124 и Arg180(по 3 контакта). Их контакты с ДНК показаны на рис.1 и рис.2. Очевидно, что аминокислотный остаток наиболее важный для распознования последовательности ДНК, должен связываться с азотистыми основаниями. Таковыми как раз являются Arg180 и Arg124обеих цепей, каждый из которых связывается с тремя разными гуанинами заданной последовательности ДНК.

Рис.1.Контакты Arg124(A) с гуанинами G54 и G19

Рис.2.Контакты Arg180(В) с G13, G51 и Arg180(A) с G4.
Предсказание вторичной структуры тРНК путем поиска инвертированных повторов
Для поиска инвертированных участков в нуклеотидных последовательностях воспользуемся программой einverted, при этом зададим параметры Gap penalty: 12, Minimum score threshold: 20, Mismatch penalty: -4 и Match score: 3. Получаем файл sequence.inv, к котором видно, что найден только акцепторный стебель, причем длиннее на одна пару, чем тот, что был найден с помощью find_pair. Результаты сравнения представлены в табл.2.
Предсказание вторичной структуры тРНК по алгоритму Зукера
Программа mfold реализует алгоритм Зукера. Воспользуемся web вариантом и обозначим параметр р=15. Третий по счету результат поиска представлен ниже на схеме.
Folding bases 1 to 75 of 1O0C:B|PDBID|CHAIN|SEQUENCE
Initial dG = -27.80
10
---- .-UC AAGC
UGGGGUA GCC \
ACCCCAU CGG G Акцепторная и D-петли
ACCG \ -- AAUG
70 20
30
.-A UUC
CCGGA \
GGCCU U Антикодоновая петля
\ - UAG
40
50
CAUUC| UUC
CGAGG \
GCUCC G T-петля
-----^ UAA
60
Табл.2.Реальная и предсказанная вторичная структура тРНК из файла 1O0C.pdb
| Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера | |
| Акцепторный стебель | 5'-902-907-3' 5'-966-971-3' Всего 6 пар | 5'-901-907-3' 5'-965-971-3' Всего 7 пар | 7 |
| D-стебель | 5'-910-912-3' 5'-923-925-3' Всего 3 пары | Не найдено | 3 из 3 реальных |
| T-стебель | 5'-949-953-3' 5'-961-965-3' Всего 5 пар | Не Найдено | 5 из 5 реальных |
| Антикодоновый стебель | 5'-937-944-3' 5'-926-933-3' Всего 8 пар | Не найдено | 5 из 8 рельных |
| Общее число канонических пар нуклеотидов | 20 | 0 | 20 |