Реконструкция деревьев по нуклеотидным последовательностям. Деревья. содержащие паралоги
1. Построение дерева по нуклеотидным последовательностям
В данном задании мы строим филогенетическое дерево бактерий, с которыми работали и в прошлых двух практикумах, но с использованием методов, использующих в качестве входных данных нуклеотидные последовательности. К таким методам относятся Neighbor-Joining, Minimum evolution, Maximum likelihood. в качестве последовательности используем 16S РНК бактерий.
В таблице 1 для каждой из рассматриваемых бактерий приведены: AC записи EMBL, в которой обнаружена последовательность 16S rRNA, координаты этой РНК в этой записи, а также то, на прямой или обратной цепи она расположена.
Таблица 1. Информация о полученных записях, содержащих последовательность 16S РНК.
| № | Название | Мнемоника | AC записи EMBL | Координаты РНК | Цепь |
| 1 | Bacillus anthracis | BACAN | AE016879 | 29129..30635 | Прямая |
| 2 | Bacillus subtilis | BACSU | AL009126 | 30279..31832 | Прямая |
| 3 | Clostridium botulinum | CLOB1 | CP000726 | 9282..10783 | Прямая |
| 4 | Clostridium tetani | CLOTE | AE015927 | 176113..177621 | Прямая |
| 5 | Enterococcus faecalis | ENTFA | AE016830 | 1018187..1019708 | Прямая |
| 6 | Finegoldia magna | FINM2 | AP008971 | 197837..199361 | Прямая |
| 7 | Geobacillus kaustophilus | GEOKA | BA000043 | 30790..32343 | Прямая |
| 8 | Lactobacillus acidophilus | LACAC | CP000033 | 59255..60826 | Прямая |
Далее вырежем нужные участки из записи EMBL командой seqret:
seqret embl:xxxxx -sask
Поместим последовательности в один fasta-файл 16S.fasta и отредактируем названия последовательностей, оставив только мнемонику видов. Создадим выравнивание отобранных белков c помощью программы muscle с параметрами по умолчанию. Получим файл align.fasta.
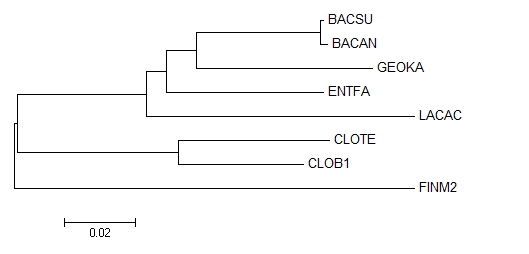
Далее построим дерево с помощью программы MEGA методом Neighbor joining (рис.1).
Рисунок 1. Дерево, построенное с помощью программы MEGA методом Neighbor joining .
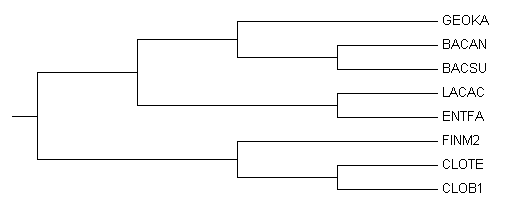
Рисунок 2. Правильное дерево.
Реконструированное дерево с правильным не совпадает.
Из пяти нетривиальных ветвей, присутствующих в правильном дереве, три присутствуют и в реконструированном по нуклеотидным последовательностям.
Присутствуют: {BACAN,BACSU} против {GEOKA,LACAC,ENTFA,FINM2,CLOTE,CLOB1}, {GEOKA,BACAN,BACSU} против {LACAC,ENTFA,FINM2,CLOTE,CLOB1}, {CLOTE,CLOB1} против {GEOKA,BACAN,BACSU,LACAC,ENTFA,FINM2}.
Отсутствуют: {LACAC,ENTFA} против {GEOKA,BACAN,BACSU,FINM2,CLOTE,CLOB1}, {FINM2,CLOTE,CLOB1} против {GEOKA,BACAN,BACSU,LACAC,ENTFA}.
2. Построение и анализ дерева, содержащего паралоги
Нашла в своих бактериях достоверные гомологи белка CLPX_BACSU. Чтобы найти гомологов в заданных организмах, воспользовалась файлом proteo.fasta на диске P, там лежат записи банка UNIPROT, относящиеся к бактериям, перечисленным в таблице к заданию 1. Провела поиск программой blastp гомологов (с порогом на E-value 0,001) и отобрала по мнемонике видов только те находки, которые относятся к отобранным мной бактериям. Сначала проиндексировала файл:
makeblastdb -in proteo.fasta -out prot -dbtype prot
Затем использовала blastp, выравнивая белки с заготовленной заранее fasta белка CLPX_BACSU.
blastp -query CLPX_BACSU.fasta -db prot -out prot_clpx.txt -evalue 0.001
Получила набор белков prot_clpx.txt и создала файл, содержащий только те находки, которые относятся к отобранным мной бактериям. Полученный файл с выравниванием импортировала в программу MEGA (использовала Maximum likelihood) и построила дерево.

Рисунок 3. Дерево, постороенное по последовательностям гомологов белка CLPX_BACSU из восьми рассматриваемых бактерий.
Два гомологичных белка будем называть ортологами, если они а) из разных организмов; б) разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования. Два гомологичных белка из одного организма будем называть паралогами.
Примеры ортологов: CLPX_GEOKA, CLPX_ENTFA, CLPX_CLOTE.
Примеры паралогов: B0S3X9_FINM2, B0S3J0_FINM2.