На данной странице представлен практикум по использованию алгоритмов blast для нуклеотидных последовательностей
1) Определение функции и таксономии нуклеотидной последовательности:
Собранную в одном из прошлых практикумов fasta последовательность ДНК ввели в качестве запроса в blastn - инструмент поиска нуклеотидной последовательности в базе нуклеотидных последовательностей. Настройки были выставлены по умолчанию, программа работала по типу blastn, чтобы поиск был наиболее чувствительным (включал в себя не только изменения белковой последовательности, но и нуклеотидные замены, не влияющие на нее), т.к. это требуется для лучшего установления филогинии и, соответственно, определения хозяина последовательности. Первые десять лучших результатов поиска доступны по ссылке.
Лучшая находка принадлежит Brada inhabilis. Поскольку у этой находки очень высокий процент идентичности с запросом (97,7%), а все несовпадающие нуклеотиды это N/W/Y, которые также совпадают с найденной последовательностью, то вероятнее всего собранная последовательность принадлежит именно этому виду.
Таксономическое положение: Eukaryota; Metazoa; Spiralia; Lophotrochozoa; Annelida; Polychaeta; Sedentaria; Canalipalpata; Terebellida; Cirratuliformia; Flabelligeridae; Brada; Brada inhabilis.
Функция белка с изучаемой последовательности описана в самой записи находки. Она кодирует гистон Н3, один из четырех белков нуклеосомы. Для проверки этой информации последовательность была введена в качестве запроса в blastx для поиска в базе данных белков. В качестве базы данных был выбран Swissprot, остальные параметры оставлены по умолчанию. Результат поиска доступен по ссылке. Из него видно, что секвенированная последовательность содержит ген белка гистона H3.
2) Поиск генов белков в неаннотированной нуклеотидной последовательности:
Для аннотации был выбран геном курицы (Gallus gallus), сборка 2021 года с АС: GCA_016699485.1. В этой сборке был выбран скэффолд с номером CM028482.1. В данном скэффолде был выбран контиг с AC: JAENSK010000010.1.
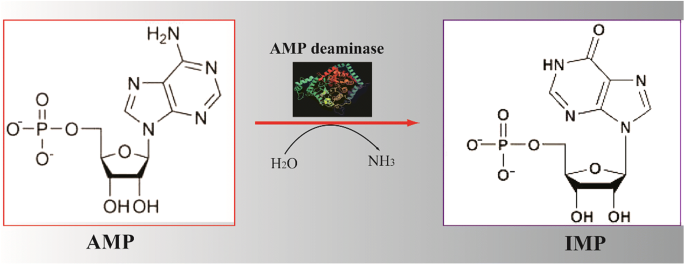
Для поиска кодирующих последовательностей в выбранном контиге соответствующий идентификационный номер был использован в качестве запроса в blastx; все параметры были выставлены по умолчанию, кроме базы белков, ограниченной белками Swiss-prot, и организма (все кроме курицы). Результат поиска доступен по ссылке. С наибольшей степенью уверенности в данном контиге обнаруживается последовательность гена АМФ-деаминазы, у которой Е-value для соответствующей найденной человеческой последовательности (Erythrocyte AMP deaminase [Homo sapiens]) равна 1e-14.
Найденный участок человеческого гена кодирует фрагмент зрелого белка АМФ-деаминазы длиной 74 аминокислоты, в то время как сам белок состоит из 767 аминокислот, т.е. процент покрытия находки примерно 10%. Этого недостаточно для корректной оценки гомологии человеческого и куриного фермента по найденным последовательностям, однако для более точной оценки нужны другие компьютерные мощности, т.к. зачастую расстояния между экзонами достигают десятков и сотен тысяч нуклеотидов. Например, в данном случае ген имеет длину 199,267 нуклеотидов, а изучаемый контиг всего 40,000 нуклеотидов, что не позволяет ему полностью покрыть последовательность белка. Однако совпадающем участке количество Identities и Positives составляют 59% и 71% соответственно, что говорит о высокой степени схожести изучаемых ферментов и, соответственно, об их вероятной гомологии. Также интересно отметить, что введенный запрос соответствует найденной последовательности не на одном, а на четырех участках (20525-20743, 11625-11843, 29463-29654, 38456-38620), что говорит о наличии повторяющихся участков в гене АМФ-деаминазы. Вероятно, это обусловлено наличием альтернативного сплайсинга для данного гена.
АМФ-деаминаза катализирует реакцию дезаминирования аденозинмонофосфата в азотистом основании по 6 положению до инозинмонофосфата. Данное вещество участвует в метаболизме азотистых оснований, а также входит в состав РНК.
3) Интепретация карты локального сходства гомологичных хромосом двух бактерий:
Для сравнения взяли геномы Mycobacterium tuberculosis (NC_000962.3) и Mycobacterium kansasii (NC_022663.1). Карта локальных свойств была построена парным выравниванием геномов изучаемых бактерий с помощью алгоритма blastn. Параметры выставлены по умолчанию.
Из картинки следует, что последовательности в целом гомологичны, но в эволюции произошли глобальные перестройки генома. Во-первых, отсутствие главных диагоналей говорит о том, что в геномах были вставки и делеции. Вставки точно были в геноме Mycobacterium kansasii, т.к. ее геном примерно на 2 Mbp длиннее другого. Во-вторых, произошли парные перестановки крупных участков в каком-либо геноме. Поменялись участки 0-1М и 2М-3М (координаты по оси X) и 1,5М-2М и 4М-4,4М. В-третьих, участки 1М-1,5М и 3М-4М инверсированы у одной из бактерий.