Профили
Построение профиля подсемейства и проверка его работы
Для работы я выбрал подсемейство из последовательностей, проанализированных в прошлом практикуме. Для этого на дереве (полученном в прошлом практикуме) я выбрал кладу, последовательности которой имеют различные архитектуры, однако все принадлежат нематодам, а точнее представителям класса Chromadorea.
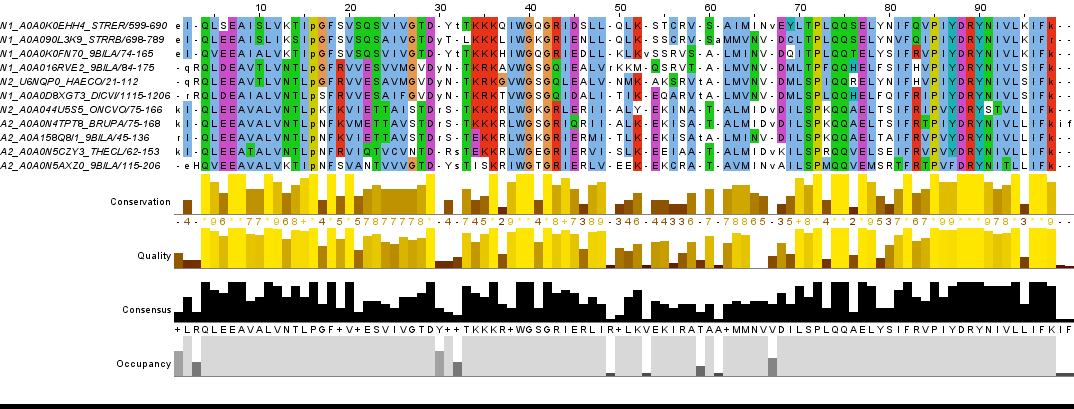
Рис. 1. Выравнивание выбранного подсемейства
Для выбранного подсемейства я построил профиль, использовав программу HMMER:
hmm2build profile.out clada.fasta
После этого, полученный профиль был откалиброван:
hmm2calibrate profile.out
Из предыдущего практикума был взят файл полученный из базы Uniprot, содержащий последовательности всех белков с рассматриваемым доменом. uniprot.txt. По данному файлу я осуществил поиск, используя откалиброванный профиль:
hmm2search profile.out uniprot.fasta > result.out
Все находки лежат в файле result.txt
Для удобства работы с данными, я скопировал первую таблицу из файла в первый лист файла Exel, в котором и производил дальнейшие обсчеты.
На листе "Представители подсемейства" я посмотрел, какие из представителей, по которым строился профиль, оказались найденными в результате. По итогу нашлись все представители, у всех очень хороший E-value. Также, все они были помечены "+" в файле Exel.
Построение ROC-кривой и гистограммы весов находок
"ROC-кривая (англ. receiver operating characteristic, рабочая характеристика приёмника) — график, позволяющий оценить качество бинарной классификации, отображает соотношение между долей объектов от общего количества носителей признака, верно классифицированных как несущих признак, (англ. true positive rate, TPR, называемой чувствительностью алгоритма классификации) и долей объектов от общего количества объектов, не несущих признака, ошибочно классифицированных как несущих признак (англ. false positive rate, FPR, величина 1-FPR называется специфичностью алгоритма классификации) при варьировании порога решающего правила." (wiki)
Для построения ROC-кривой я посчитал следующие параметры: TP (True Positives) - количество последовательностей, расположенные выше некоторого порога (т.е. на некотором уровне значимости содержащих искомый домен); TN (True Negatives) - обратное, т.е. кол-во последовательностей достоверно не содержащих искомый домен; FP (False Positives) - ошибки II рода, т.е. кол-во последовательностей расположенных выше порога, но не содержащих домен; FN (False Negatives) - ошибки I рода, т.е. количество последовтельностей расположенных ниже порога, но содержащих домен (ложный пропуск); SP (специфичность), SP=TN/(TN+FP) - доля достоверно предсказанных белков, не содержащих домен, от общего количества последовательностей, известно не содержащих домен; SE (чувствительность), SE=TP/(TP+FN) - доля достоверно предсказанных белков, содержащих домен, от общего количества последовательностей, известно содержащих домен
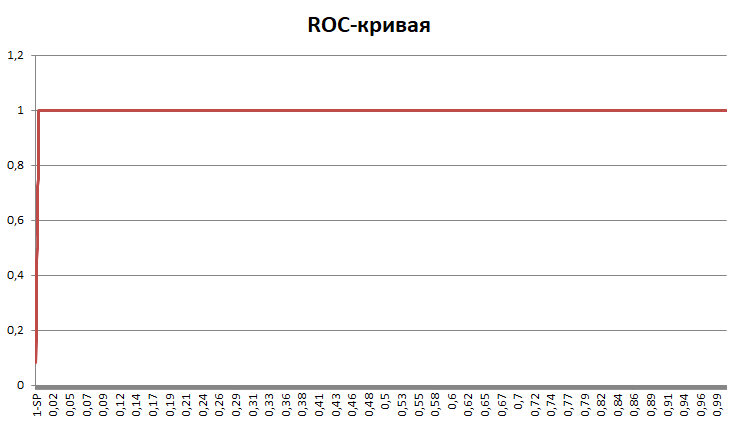
Рис. 2. ROC-кривая
Максимальное значение SP+SE-1 отражает состояние, когда SP и SE одновременно максимальны. Основываясь на этом, можно выбрать порог достоверности - 111,2 (E-value = 1,4E-30).
Также я построил гистограмму весов находок:
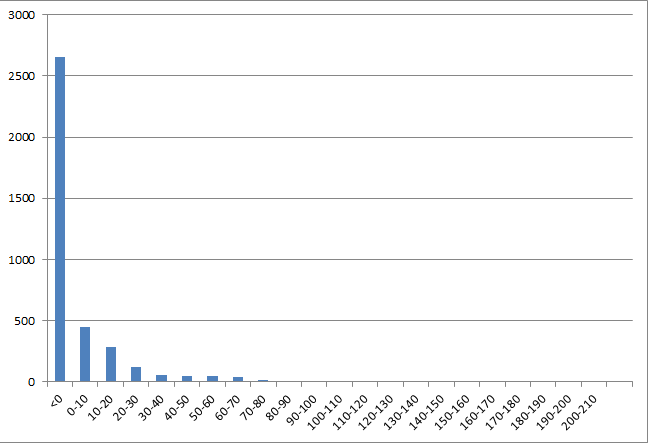
Рис. 3. Гистограмма весов всех находок
Мне показалось, что будет более информативно, если добавить гистограмму без отрицательных весов, с более сгруппированными большими значениями:
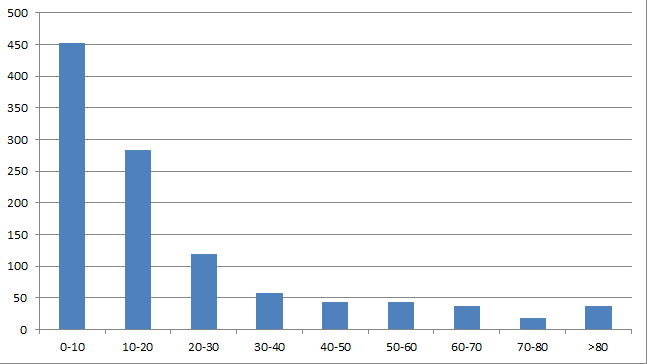
Рис. 4. Гистограмма положительных весов находок
Таблица 1. Анализ данных порогом достоверности
| На самом деле | принадлежит подсемейству | не принадлежит | сумма |
| Выше порога по профилю | 11 | 20 | 31 |
| Ниже порога | 0 | 3963 | 3963 |
| сумма | 11 | 3983 | 3994 |
| на главную |
© Гавриш Глеб 2017 |