EMBOSS. Выравнивание геномов.
Задание 1. Oтчёт о выполнении упражнений пакета EMBOSS.
| Отчетная таблица | |||
|---|---|---|---|
| Задание (программа) | Скрипт | Входные файлы | Итоговые файлы |
| 1. seqret - Несколько файлов в формате .fasta собрать в единый файл. | seqret "\*_HUMAN.fasta" 3_HUMAN.fasta | CISY_HUMAN.fasta; HSP7C_HUMAN.fasta; RPB2_HUMAN.fasta | 3_HUMAN.fasta |
| 2. seqretsplit - Один файл .fasta разделить на несколько fasta-файлов. | seqretsplit 3_HUMAN.fasta | 3_HUMAN.fasta | o75390.2.fasta; p11142.1.fasta; p30876.1.fasta |
| 3. seqret - Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta-файле. | echo -e "gb::genbank: CP016975[634:2274]\n gb::genbank: CP016975[3878:5032]\n gb::genbank: CP016975[6822:8447:r]" >cds2.txt seqret @cds2.txt cds2.fasta | CP016975.1 | Промежуточный: cds2.txt Итоговый: cds2.fasta |
| 4. transeq - Транслировать кодирующие последовательности, лежащие в одном fasta-файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta-файле. | ["/transl_table=11" для Brucella canis]
transeq -table 11 cds.fasta trans_cds.fasta | cds.fasta | trans_cds.fasta |
| 5. transeq - Транслировать данную нуклеотидную последовательность в шести рамках. | transeq -frame 6 cds2.fasta trans6_cds.fasta cat trans6_cds.fasta | cds2.fasta | trans6_cds.fasta |
| 6. seqret - Перевести выравнивание и из fasta формате в формат .msf. | seqret panthera_align.fasta msf::panthera_align.msf | panthera_align.fasta | panthera_align.msf |
| 7. infoalign - Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число). | infoalign panthera_align.fasta -refseq 2 -only -name -idcount stdout | panthera_align.fasta |  |
| 8. featcopy - Перевести аннотации особенностей в записи формата .gb в табличный формат .gff. | featcopy brucella.gb brucella.gff | brucella.gb | brucella.gff |
| 9. extractfeat - Из данного файла с хромосомой в формате .gb получить fasta-файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product). | extractfeat seq2.gb info.fasta -type CDS -describe product | seq2.gb | info.fasta |
| 10. shuffle - Перемешать буквы в данной нуклеотидной последовательности; (*) проверить с помощью
blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных (с порогом E = 10 - по умолчанию). P.S. ИЗ-ЗА ТЕХНИЧЕСКИХ РАБОТ НА САЙТЕ NCBI ПОКА НЕ УДАЕТСЯ ВЫПОЛНИТЬ ПРОВЕРКУ. P.P.S. Пришлось поменять организм из-за ошибки программы. |
shuffle -o shuffled_cds.fasta cds.fasta | cds.fasta | shuffled_cds.fasta |
| 11. cusp - Найти частоты кодонов в данных кодирующих последовательностях. | cusp cds.fasta freq.cusp | cds.fasta | freq.cusp |
| 12. compseq - Найти частоты динуклеотидов в данной нуклеотидной последовательности и сравнить их с ожидаемыми. | compseq -word 2 -calcfreq cds.fasta cds.compseq | cds.fasta | cds.compseq |
| 13. tranalign - Выровнять кодирующие последовательности соответственно выравниванию белков (их продуктов). | tranalign -asequence nucls.fasta -bsequence pepts.fasta -outseq nucls_align.fasta | nucls.fasta; pepts.fasta | nucls_align.fasta |
Задание 2. Описание глобальных эволюционных событий и определение сходства гомологичных участков ДНК для
полных геномов.
2b. Построение нуклеотидного пангенома для 3 геномов близкородственных бактерий.
| Геномы близкородственных бактерий содержат высокосходные последовательности ортологичных
фрагментов, но обычно претерпевают множественные перестройки длинные делеции, вставки мобильных элементов и иногда горизонтально перенесенные участки. Программа
Nucleotide PanGenome explorer (NPG-explorer) предназначена для выравнивания и анализа множества близкородственных геномов. NPG-explorer создает нуклеотидный
Пангеном - множество выровненных блоков, состоящих из ортологичных фрагментов. Фрагменты, у которых нет ортологов, считаются вырожденными блоками из одного
фрагмента. Каждый нуклеотид из входных геномов принадлежит ровно одному блоку нуклеотидного пангенома. Параметры алгоритма: минимальная длина блока (по умолчанию 100 нуклеотидов) и минимальная идентичность (по умолчанию 90%). NPG-explorer повторяет алгоритм нахождения блоков, пока следующий критерий не будет удовлетворён: поиск BLAST всех против всех не находит новых блоков достаточной длины и идентичности. NPG-explorer выдаёт следующие данные:
Аналитические файлы с полезной информацией. [1] Используемые определения: Пангеном — суммарный набор генов каждого вида, который можно подразделить на три части: универсальные гены (есть у всех штаммов), периферические гены (есть у большей части штаммов) и штамм-специфичные, уникальные, гены. Пангеном может быть построен на основе ортологичных генов или ортологичных участков генома (нуклеотидный пангеном); Кор — блоки, которые включают участки, встречающиеся во всех геномах; Дополнительные участки — встречаются не во всех геномах; Уникальные участки — встречаются только в одном геноме. pangenome/pangenome.info содержит сводную информацию про все типы блоков:
pangenome/pangenome.bi содержит информацию по каждому блоку, включая информацию о том, фрагменты каких геномов входят в блок; удобен для поиска крупных инделей (h-блоки и u-блоки) и анализа блоков с повторами. Список глобальных блоков - синтений - см. в global-blocks/blocks.gbi. g-блоки (глобальные блоки) состоят из последовательно идущих во всех геномах s-блоков, перемежающихся блоками других типов (r-, h-, u- и m-). Последовательность глобальных блоков в каждом геноме см. в файле global-blocks/blocks.blocks. [2] |
Для построения нуклеотидного пангенома были выбраны штаммы Bacillus subtilis HJ5, PS832, SG6.
Bacillus subtilis (cенная палочка) — вид грамположительных спорообразующих аэробных бактерий, представителей рода бациллы (Bacillus).
Bacillus subtilis — один из наиболее хорошо изученных микроорганизмов. Название сенная палочка происходит из-за того, что ранее Bacillus subtilis изолировался
исключительно из сенных отваров. Bacillus subtilis имеет вид бесцветной прямой палочки, размером примерно 0,7 мкм в толщину и 2—8 мкм в длину. Может размножаться делением
и спорами. Иногда отдельные представители, после поперечного деления, остаются соединенными в нити. Благодаря продуцируемым антибиотикам и способности закислять среду
обитания, является антагонистом патогенных и условно-патогенных микроорганизмов, таких как сальмонелла, протей, стафилококки, стрептококки, дрожжевые грибки; продуцирует
ферменты, удаляющие продукты гнилостного распада тканей; синтезируют аминокислоты, витамины и иммунноактивные факторы. [3]
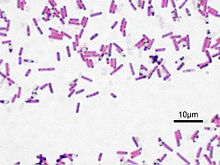
Bacillus subtilis, окраска по Граму
1. Алгоритм построения нуклеотидного пангенома.
С помощью программы NPG-explorer, установленной на сервере kodomo, был построен пангеном [название бактерий]. Рабочая директория проекта:gladmarine/term3/block2/pr9_(pr7_emboss_genome_align)/bac_subt_npgВ ней находится основной файл - genomes.tsv, куда я записала ссылки на последовательности исследуемых штаммов бактерий.
В результате работы команды
npge Prepare(скачивает и переименовывает геномные ДНК) были созданы следующие файлы: genomes-renamed.fasta - fasta-файл с последовательностями геномов для построение нуклеотидного пангенома и genes/features.bs - собрание блоков генов (1 ген представлен в качестве 1 блока). Файлы genomes-raw.fasta и features.embl содержат необработанную исходную информацию и не используются в дальнешей работе, поэтому могут быть удалены.
После
npge Examineв специально созданную директорию examine записываются файлы: genomes-info.tsv - таблица длин геномов; draft.bs - шаблон пангенома; identity_recommended.txt - важный текстовый файл с рекомендуемым значением параметра MIN_IDENTITY (0.879, его я указала вместо дефолтного 0.9) в файле npge.conf
npge -g npge.confСам пангеном был результатом команды
npge MakePangenome,которая создает файл в BlockSet-формате pangenome/pangenome.bs, а большой список файлов форматов .bs (содержат блоксэты), .bi (содержат таблицы характеристик блоков), .ba (с выравниваниями блоксэтов, ячейки - фрагменты), .blocks (с выравниваниями блоксэтов, ячейки - блоки) с полной информацией о построенном пангеноме был вызван посредством
npge PostProcessing.
2. Описание синтеничных участков.
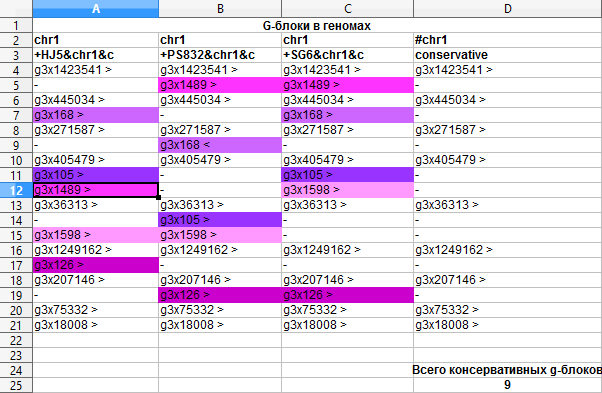
Данное задание предполагало анализ g-блоков. Для этого содержимое файлов blocks.gbi (с описанием синтенией) и blocks.blocks (с последовательностью глобальных блоков в каждом геноме) было импортировано в Excel. Таблица транспонирована и раскрашена по блокам. Строки, вообще не содержащие g-блоков (i-blocks), удалены. Выравнивание в qnpge не отличается от табличного варианта, поэтому не было приведено.Всего было найден 18 блоков, половина из которых оказалась консервативными.
Можно сказать, что попарные выравнивания имеют примерно одинаковый вес. Так при сравнении HJ5 и SG6 находим 2 позиции с точно совпадающими значениями - g3x168> и g3x105>, у PS832 и SG6 их также 2 - g3x1489> и g3x126>. Поскольку некоторые блоки есть во всех трёх последовательностях, но в двух из них их позиция совпадает, а в третьей отличается, можно рассуждать о транслокации блоков, при этом, характер перемещения составляет 2 позиции, вероятно, из-за кольцевой формы хромосомы. Любопытен тот факт, что ориентация всех блоков в данных геномах совпадает, то есть инверсий не происходило. Кроме того, стоит отметить транслокацию(?) g-блока g3x1489> у штамма HJ5 относительно расположения этого блока во второй позиции у других штаммов.
Таблица g-блоков
3. Описание ядра пангенома.
Ядро пангенома представляет собой объединение s-блоков, присутствующих во всех геномах. Информация о них содержится в ~/pangenome/pangenome.info в секции "Exact stem blocks".- Число s-блоков: 218
- Размер ядра (процент входных последовательностей, вошедших в s-блоки - "Total length of blocks"): 3717425 (81.81%)
- Сходство геномов (в объединенном выравнивании - "Identity of joined blocks"): 0.978228
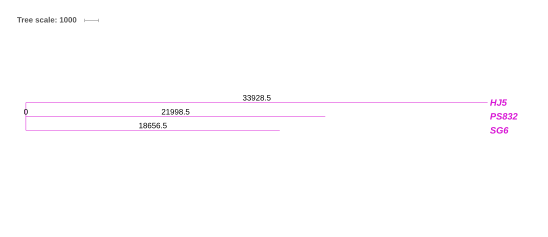
4. Филогенетическое дерево.
Построено по объединенному выравниванию s-блоков (nj-global-tree.tre) с использованием программы iTOL. Судя по длине ветвей, более родственными являются штаммы PS832, SG6, являющиеся потомками штамма HJ5 (что я и предполагала, основываясь на данных проанализированной выше таблицы). Длины обычно обозначают отношение в процентах общего число замен нуклеотидов к длине последовательности. [4]. В данном случае (как мне кажется) указаны длины (из ~/trees/nj-global-tree-full.tre) консервативных позиций.Филогенетическое дерево
5. Описание повторов.
- Число r-блоков: 460
- Суммарная длина (процент от средней длины генома - "Total length of blocks"): 27227 (0.59%)
- Сходство геномов (в объединенном выравнивании - "Identity of joined blocks"): 0.944097
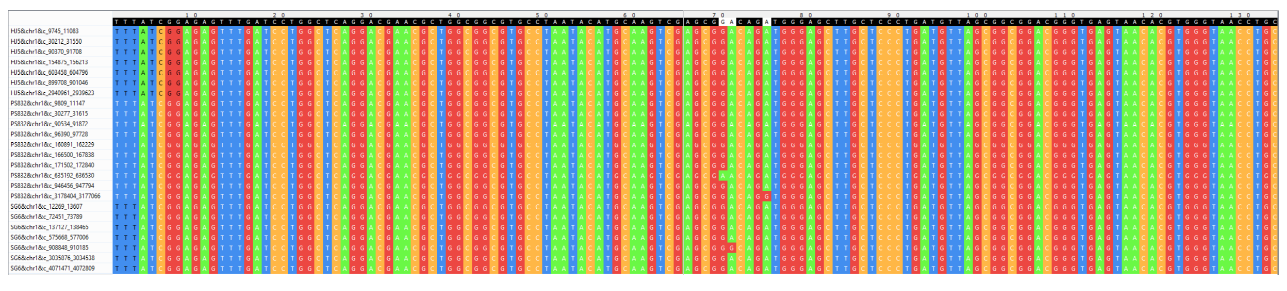
Для анализа был выбран блок r24x1339, состоящий из 24 фрагментов с общей длиной 1339 нуклеотидов. В геномах HJ5 и SG6 его повторы встречаются 7 раз, у штамма PS832 их 10. Процент идентичности составляет более 99%, что говорит о сильной консервативности, следовательно, есть смысл ожидать кодирующую последовательность в выдаче BLAST. По предсказанию qnpge данный фрагмент кодирует 23S рибосомальную РНК [стоит проверить по BLAST].
Фрагмент исследуемого r-блока
6. Пример крупной делеции. H-блоки.
- Число h-блоков: 160
- Суммарная длина (процент от средней длины генома - "Total length of blocks"): 146656 (3.22%)
- Сходство геномов (в объединенном выравнивании - "Identity of joined blocks"): 0.977959
Информация из qnpge

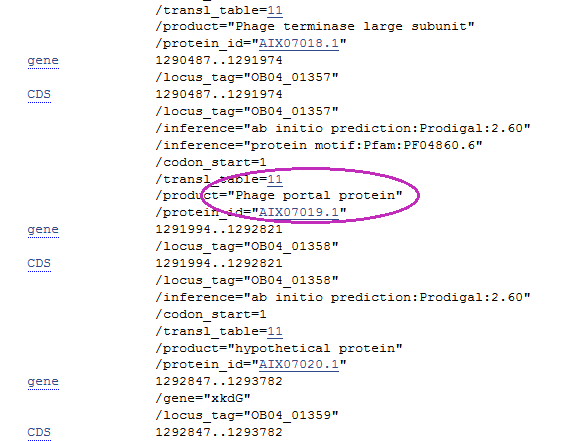
С помощью проверки по базе NCBI было выявлено, что phage portal protein также кодируется в SG6, однако располагается в другом локусе. Скорее всего, можно предполагать транслокацию, нежели делецию.
Сраница SG6 в NCBI
7. Пример последовательности, имеющейся только в одном геноме.
В качестве примера был выбран блок u1x3231 штамма HJ5. На данном участке qnpge определил 3 гена, для одного из которых были также определены рамки считывания:- ген, кодирующий oligopeptide ABC transporter permease, 915 bp >, старт-кодон ATG (п.1557-1559), стоп-кодон TAA (п.2505-2507)
- ген, кодирующий nucleic acid binding protein, 915 bp >, старт-кодон ATG (п.2524-2526)
- ген, кодирующий oligopeptide ABC transporter permease, 1629 bp >, стоп-кодон TAA (п.1476-1478)