Практикум №13. Ресеквенирование и поиск полиморфизмов у человека.
Ресеквенирование. Поиск полиморфизмов у человека.
Часть 1. Подготовка чтения.
Для задания была взята 16-ая хромосома.
Для начала при помощи команды fastqc chr16.fastq был создан архив с данными о прочтении.
Также был создан html-файл, в котором содержится отчет о проделанной программой работе.
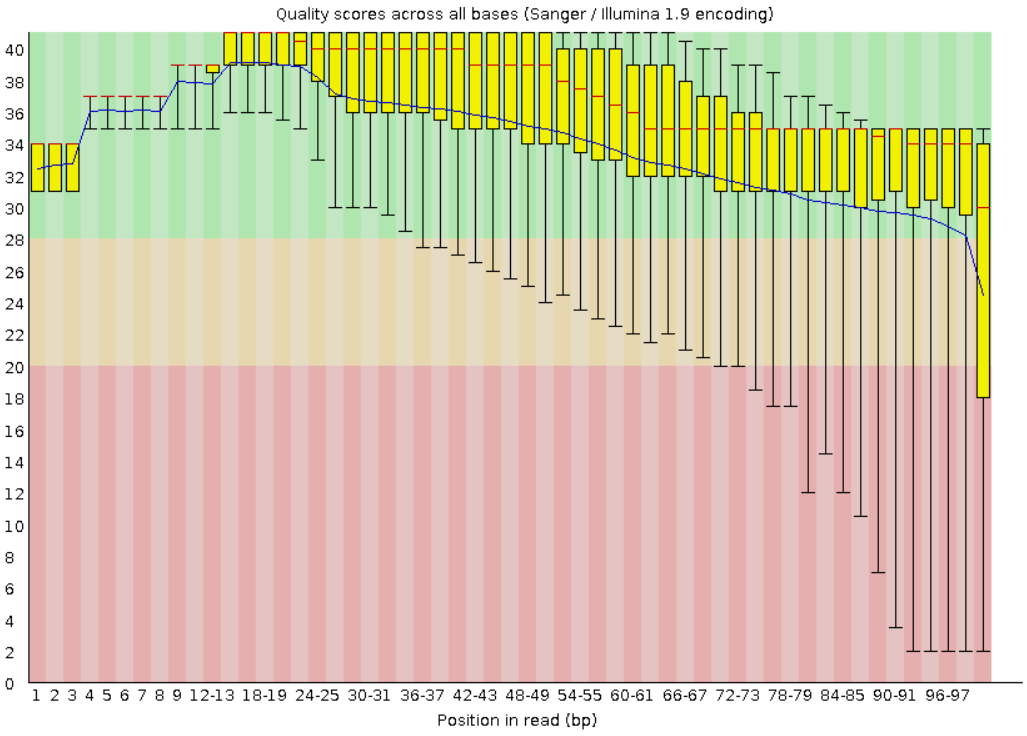
Рис.1 Качество нуклеотидов в чтениях до очистки. Число чтений 3965.
Далее требовалось провести очистку чтений от участков с плохим качеством.
Для этого использовались следующие команды:
- java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr16.fastq trim1.fastq LEADING:20 - для удаления участков со скором меньше 20 из начала чтений;
- java -jar /usr/share/java/trimmomatic.jar SE -phred33 trim1.fastq trim2.fastq TRAILING:20 - для удаления участков со скором меньше 20 из конца чтений;
- java -jar /usr/share/java/trimmomatic.jar SE -phred33 trim2.fastq trim3.fastq MINLEN:50 - для удаления чтений длиной менее 50.
К полученному файлу была применена команда fastqc trim3.fastq: результат
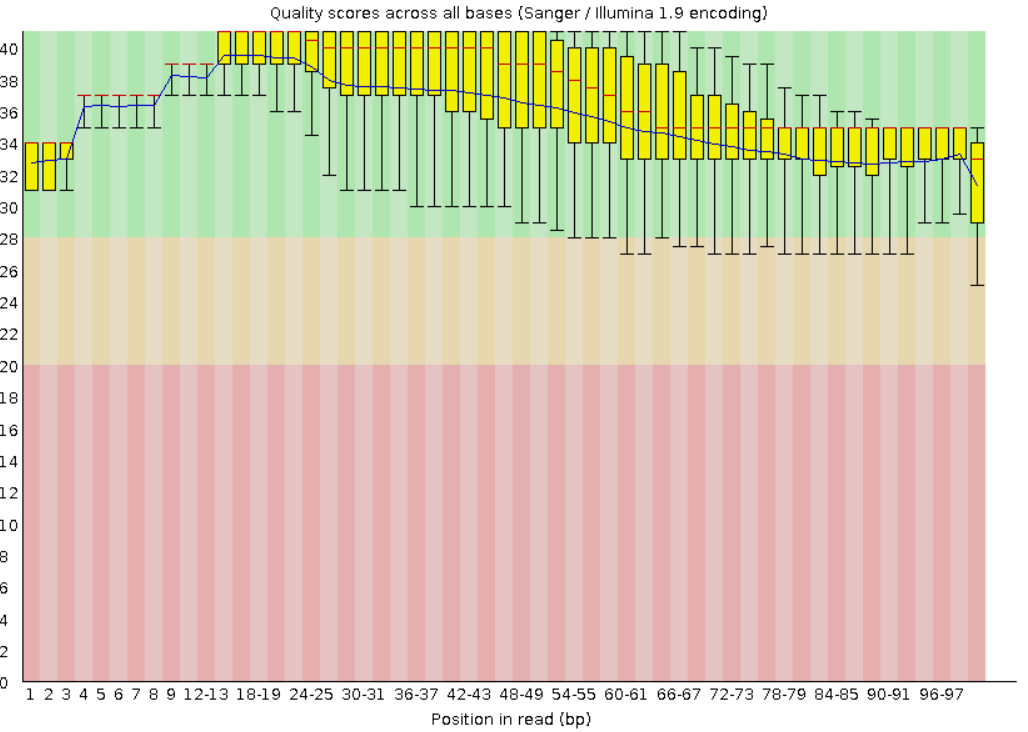
Рис.2 Качество нуклеотидов после очистки. Число чтений 3796
Часть 2. Картирование чтений.
Очищенные чтения были картирован с помощью программы BWA. Сначала рeференсная последовательность была проиндексирована с помощью команды bwa index:
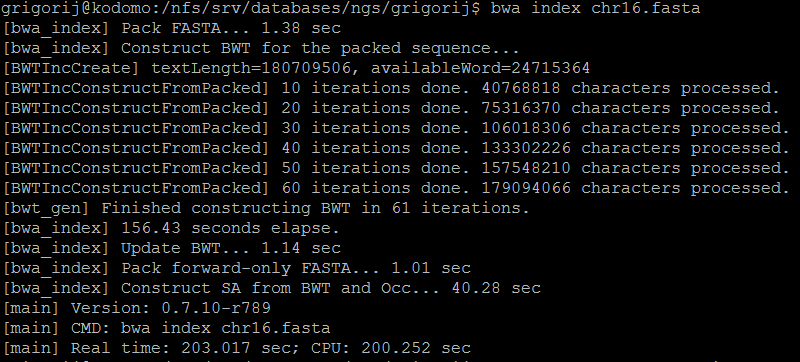
Рис.3. Результат bwa index.
Затем было произведено выравнивание референса с прочтениями:
bwa mem chr16.fasta trim3.fastq > align.sam. Полученный файл в формате sam: align.sam
Проанализируем полученное выравнивание:
- samtools view -b align.sam -o aln.bam - Переведём его в бинарный формат;
- samtools sort aln.bam out.predix - отсортируем по координате в референсе начала чтения;
- Наконец, проиндексируем полученный файл: samtools index out.predix.bam, после чего мы, наконец, сможем узнать, сколько ридов было откартировано:

Рисю4 Число картированных ридов выделено синим, некартированных красным.
Часть 3. Анализ SNP.
Для поиска SNP и инделей использовались следующие команды:
- samtools mpileup -uf chr16.fasta out.predix.bam > snp.bcf - создадим файл с полиморфизмами в формате .bcf
- bcftools call -cv snp.bcf > snp.vcf - получаем файл со cписком полиморфизмов.
Всего было найдено 63 snp и 2 инделя. Из snp.vcf выбраны следующие полиморфизмы:
- Полиморфизм с координатами 11348246 - индель (замена АСАСТСАСТС на АСАСТС), качество чтения - 10.8393, глубина - 5;
- Полиморфизм с координатами 11348273 - замена Т на А, качество чтения 4.12853, глубина 11;
- Полиморфизм с координатами 11361895 - замена C на G, качество чтения 225.009, глубина 50.
- perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp2.vcf > converted.avinput - перевод файла snp2.vcf в формат, с которым может работать annovar.
- perl /nfs/srv/databases/annovar/annotate_variation.pl -out RefGeneAnnotation -build hg19 converted.avinput //nfs/srv/databases/annovar/humandb/ - аннотация содержимого файла convert.avinput по базе данных RefGene (директория humandb) с помощью скрипта annotate_variation.pl (в директории annovar). Выдача представлена в двух формах: в файле variant_function собраны все SNP, а в exonic_variant_function - только SNP из экзонов.
- perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out SNP138Annotate -build hg19 -dbtype snp138 converted.avinput /nfs/srv/databases/annovar/humandb/ - аннотирует содержимое файла converted.avinput по базе данных snp138 из директории humandb с помощью скрипта annotate_variation.pl, находящемся в директории annovar. Проанализировав выдачу, можно узнать, что 57 snp имеют rs.
- perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_eur -buildver hg19 -out 1000gAnnotate converted.avinput /nfs/srv/databases/annovar/humandb/ - аннотирует содержимое файла converted.avinput по базе данных 1000 genomes с помощью скрипта annotate_variation.pl.
- perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out gwasAnnotate -dbtype gwasCatalog converted.avinput /nfs/srv/databases/annovar/humandb/ - аннотирует содержимое файла converted.avinput по базе данных Gwas с помощью скрипта annotate_variation.pl.
- perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 converted.avinput -outfile snp_clinvar /nfs/srv/databases/annovar/humandb/ - аннотирует содержимое файла converted.avinput по базе данных Clinvar с помощью скрипта annotate_variation.pl.
- 1000 genomes - Общая информация по полиморфизмам;
- RefGene - Ген, в котором находится конкретный полиморфизм;
- snp138 - Собственно, база snp;
- Gwas - Связь между полиморфизмом и заболеванием;
- Clinvar - Возможно, связь между полиморфизмом и фенотипом (в данном случае совпадений не было найдено).
Далее я аннотировал все SNP, предварительно удалив все(2) индели, т.к. они нас не интересуют. Для этого использовалась программа annovar:
После аннотации были получены следующие базы данных:
Разброс частоты найденных SNP - от 14% до 99%
Категории на к-рые делятс SNP по RefGene: downstream, exonic, intergenic, intronic, upstream, UTR3, UTR5. Мутации попали в категории: intronic и exonic.
Гены в которые попали полиморфизмы - HERPUD1, PRM1, PRM2, PRM3, RMI1, RMI2, PRSS53, TNP2, SOCS1.
Была создана сводная таблица, которая содержит общую информацию (лист полиморфизмы) и данные о заменах аминокислот(лист замены АК).
Из всех данных можно сделать вывод, что пациент возможно имеет склонность к ожирением (частота 0.68) и имеет предрасположенность к метаболическому синдрому (частота 0.14) и к болезне Паркинсона (частота 0.37).