Поиск полиморфизмов у пациента
Часть I. Подготовка чтений
Для работы мне был выдан файл с одноконцевыми чтениями экзома 5 хромосомы человека chr5.fastq, и файл с последовательностью этой хромосомы из генома со сборкой версии hg19 chr5.fasta.
Сперва я выполнила анализ качества чтений с помощью программы FastQC. Я воспользовалась версией, установленной на сервере kodomo. В командной строке я написала:
fastqc chr5.fastq
После чего программа в той же папке выдала мне файл chr5_fastqc.html, содержащий визуализированный отчёт о работе, и архив chr5_fastqc.zip с каринками и текстовыми файлами.
Следующим шагом я выполнила очистку чтений с помощью программы Trimmomatic. Почитав руководство пользователя я нашла операции, с помощью которых можно отрезать с конца каждого чтения нуклеотиды с качеством ниже 20 и оставить только чтения длиной не меньше 50 нуклеотидов. Ниже представлена подробная информация о выбранных мною операциях.
- TRAILING - удаляет с конца чтения нуклеотиды с качеством ниже указанного. Например, следующая запись означает удаление нуклеотидов с качеством ниже 20.
TRAILING:20
Как только следующий нуклеотид окажется с Q > 20 или = 20, программа приступет к следующему чтению.
MINLEN:50
Удалённые чтения будут подсчитанны и включены в графу "опущенные чтения", представленную в итоге работы программы.
Я воспользовалась версией программы Trimmomatic, установленной на сервере kodomo. В командной строке я писала следующую команду:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr5.fastq chr5_out.fastq TRAILING:20
MINLEN:50
В результате я получила очищенный файл chr5_out.fastq, который я проанализировала с помощью программы FastQC. Для этого я ввела в командную строку:
fastqc chr5_out.fastq
На выходе я получила файл с отчётом chr5_out_fastqc.html.
Теперь выполним сравнение чтений до и после очистки.
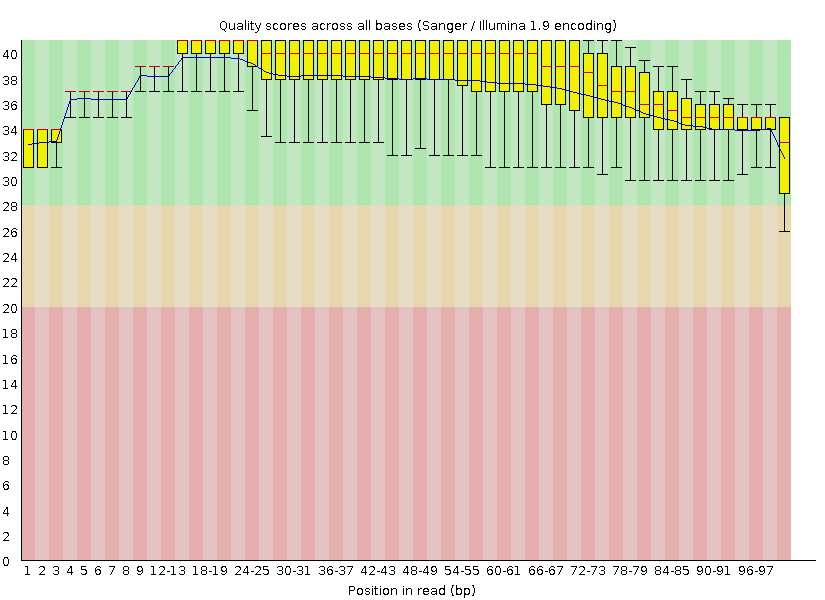
1) До очистки. Число чтений = 8208. Длина чтений = 41-100. Процен GC = 37%.
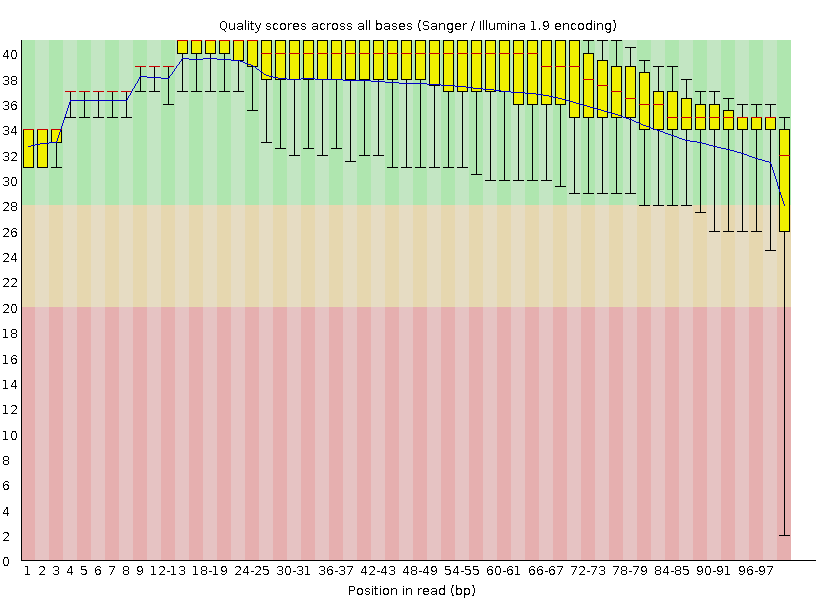
2) После очистки. Число чтений = 8114. Длина чтений = 50-100. Процен GC = 37%.
Часть II. Картирование чтений
Сперва было необходимо проиндексировать референсную последовательность. Для этого я воспользовалась программой BWA (Burrows-Wheeler Aligner). Так как она установле на сервере kodomo, то я запустила её, введя в командной строке:
bwa index chr5.fasta
В результате работа программы заняла почти 6 минут (342.318 секунд). Затем я построила выравнивание очищенных прочтений и референса в формате .sam по алгоритму mem. Для этого я выполнила команду:
bwa mem chr5.fasta chr5_out.fastq > chr5.sam
Работа программы заняла 3.5 секунд, и на выходе я получила файл chr5.sam. Теперь мне нужно было перевести его в бинарный формат .bam. Для этого я воспользовалась следующей командой пакета samtools:
samtools view chr5.sam -bo chr5.bam
Опция -b меняет формат файла на .bam, а опция -o указывает на название выходного файла. В результате я получила файл chr5.bam. Затем я отсортировала получившийся файл по координате начала чтения в референсе. Для этого я использовала команду:
samtools sort chr5.bam -T t.txt -o chr5_sort.bam
Опция -T позволяет записывать временные файлы в t.txt, а не в stdout, а опция -o указывает на название выходного файла. В результате я получила отсортированный файл chr5_sort.bam. Затем было необходимо его проиндексировать, для чего я воспользовалась командой:
samtools index chr5_sort.bam
В результате я получила индексный файл chr5_sort.bam.bai. Последней задачей по работе с пакетом samtools было выяснить, сколько чтений откартировалось на геном. Для этого я воспользовалась командой:
samtools idxstats chr5_sort.bam > out.txt
В результате я получила файл out.txt в котором указаны название последовательности, длина последовательности (180915260) и число картированных чтений (8113). Получается, что лишь одно чтение не было картировано.
Часть III. Анализ SNP
Прежде всего я создала файл с полиморфизмами в формате .bcf, для чего воспользовалась командой:
samtools mpileup -uf chr5.fasta chr5_sort.bam -o chr5.bcf
Здесь опция -u создаёт несжатый выходной файл, а опцией -f мы задаём референсную последовательность. На выходе я получила файл chr5.bcf, который затем использовала для создания файла со списком отличий между референсом и чтениями в формате .vcf с помощью следующей команды из пакета bcftools:
bcftools call -cv chr5.bcf -o chr5.vcf
В результате я получила файл chr5.vcf. Проведём его анализ.
- Всего обнаружено 32 полиморфизма
- 2 вставки
- 2 делеции
- 28 замен (SNP)
В таблице 1 представлены данные из файла chr5.vcf о наиболее интересных полиморфизмах.
| Таблица 1. Описание некоторых полиморфизмов | |||||
| тип | координата (chr5) |
референс | чтение | глубина покрытия | качество чтений |
| вставка | 35857308 | T | TC | 45 | 178.458 |
| вставка | 74639576 | C | CAA | 36 | 217.468 |
| делеция | 74639544 | CTTGTATTGT | CTTGT | 30 | 73.4665 |
| делеция | 74652239 | CAG | C | 5 | 58.0073 |
| замена | 74633975 | C | T | 1 | 8.64911 |
| замена | 35874575 | C | T | 164 | 225.009 |
Прежде, чем перейти непосредственно к аннотации SNP (однонуклеотидных полиморфизмов), я удалила все индели (делеци + вставки) из файла chr5.vcf и получила файл chr5_new.vcf. Затем я воспользовалась скриптом convert2annovar.pl, который конвертировал мой файл в формат ANNOVAR:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr5_new.vcf -outfile chr5.annovar
Далее с полученным файлом chr5.annovar можно произвести аннотации SNP с помощью скрипта annotate_variation.pl.
Аннотация в RefGene
Версия: refGene
Тип аннотации: gene-based annotation (основана на генной разметке)
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr5.refgene -build hg19 chr5.annovar
/nfs/srv/databases/annovar/humandb/
В результате я получила файлы chr5.refgene.exonic_variant_function (таблица с описанием полиморфизмов, попавших в экзоны), chr5.refgene.variant_function (таблица с описанием расположения полиморфизмов относительно генов), chr5.refgene.log (отчёт о работе команды). База данных RefGene делит все SNP на категории, перечисленные в таблице 2 с указанием количества попавших в эти участки исследуемых полиморфизмов.
| Таблица 2. Зоны расположения SNP | ||
| Категория | Описание | Полиморфизмы из chr5 |
| exonic | SNP в кодирующей области гена (экзоне) | 4 |
| splicing | SNP в пределах 2 нуклеотидов со стороны интрона от места соединения при сплайсинге (порог можно изменить) | 0 |
| ncRNA | SNP в транскрипте, не имеющем аннотацию кодирующего участка (на самом деле транскрипт может являться кодирующим) | 0 |
| UTR5 | SNP в 5'-нетранслируемой области | 0 |
| UTR3 | SNP в 3'-нетранслируемой области | 1 |
| intronic | SNP в некодирующей области гена (интроне) | 23 |
| upstream | SNP в пределах области из 1000 нуклеотдов до сайта начала транскрипции (порог можно изменить) | 0 |
| downstream | SNP в пределах области из 1000 нуклеотдов после сайта конца транскрипции (порог можно изменить) | 0 |
| intergenic | SNP в межгенной области | 0 |
16 полиморфизмов оказались гомозиготными, а 12 - гетерозиготными. Также в выходном файле указаны названия генов, в которые попали полиморфизмы. В моём случае это гены:
- IL7R - ген, кодирующий белок - альфа-цепь рецепторов интерлейкина 7 и TSLP. Они встраиваются в наружние мембраны клеток имунной системы. Рецептор интерлейкина 7 обнаружен в B-клетках и Т-клетках, а также в их предшественниках. Рецептор TSLP найден в В-клетах, Т-клетках, моноцитах и дендритных клетках. Связывание рецепторов с соответствующими цитокинами влияет на регуляцию активности клеток имунной системы.
- CAPSL - ген, кодирующий белок, связывающий ионы кальция (Calcyphosin-like protein). Он очень похож по структуре на белок Calcyphosin, за что и получил своё название. Оба этих белка, возможно, играют роль в регуляции ионного транспорта. Однако экспериментально их функции плохо изучены.
- HMGCR - ген, кодирующий трансмембранный белок - 3-гидрокси-3-метилглютарил-CoA редуктазу (HMG-CoA reductase). Это фермент, катализирующий лимитирующую стадию синтеза холестерола (синтез мевалоновой кислоты). Он регулируется по механизму отрицательной обратной связи. HMG-CoA редуктаза является основной мишенью лекарственных препаратов, снижающих уровень холестерина.
Интерлейкин 7 (IL-7) - цитокин, фактор роста клеточных компонентов крови, важен для развития B-клеток и T-клеток, стимулирует дифференциацию стволовых клеток в предшественников лимфоцитов.
Thymic stromal lymphopoietin (TSLP) - цитокин эпителиальных клеток, который регулирует дифференциацию Т-клеток.
Так как наиболее значимыми являются нуклеотидные замены в кодирующих областях генов, которые могут приводить к замене одного аминокислотного остатка в белке на другой, то рассмотрим подробнее выходные данные о полиморфизмах в экзонах в таблице 3.
| Таблица 3. Описание полиморфизмов в экзонах | ||||||||
| ген | координата (chr5) |
референс | чтение | тип замены |
синонимичность замены |
глубина покрытия |
качество чтений |
аминокислотная замена |
| IL7R | 35861068 | T | С | гомозиготная | нет | 44 | 221.999 | I66T |
| IL7R | 35871190 | G | А | гомозиготная | нет | 98 | 221.999 | V138I |
| IL7R | 35874575 | С | Т | гетерозиготная | нет | 164 | 225.009 | T244I |
| CAPSL | 35910529 | С | Т | гомозиготная | нет | 92 | 221.999 | R85Q |
Комментарий к таблице 3:
- I66T - замена 66 остатка гидрофобного изолейцина на остаток гидрофильного треонина. Возможно изменение пространственной структуры белка. Однако эта замена является естественной и часто встречающейся.
- V138I - замена 138 остатка валина на схожий по свойствам остаток изолейцина. Маловероятны существенные изменения в работе белка. Эта замена является естественной и часто встречающейся.
- T244I - замена 244 остатка гидрофильного треонина на остаток гидрофобного изолейцина. Возможно изменение пространственной структуры белка. Интересно, что присутствие на 244 позиции остатка треонина учёные связывают с предрасположенностью пациента к рассеянному склерозу. А полиморфизм с изолейцином считается нормой, хотя и менее распространённой.
- R85Q - замена 85 остатка гидрофильного, положительно заряженного аргинина на остаток гидрофильного глутамина. Возможно лишь незначительное изменение пространственной структуры белка, так как замена не попала в каталитические центры (52-63 и 88-99 аминокислотные остатки).
Аннотация в dbSNP (Single Nucleotide Polymorphism Database)
Версия: snp138
Тип аннотации: filter-based annotation (основана на фильтрации)
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr5.dbsnp -build hg19 -dbtype
snp138 chr5.annovar /nfs/srv/databases/annovar/humandb/
В результате я получила файлы chr5.dbsnp.hg19_snp138_dropped (перечень полиморфизмов, аннотированных в базе данных dbSNP, имеющих rs), chr5.dbsnp.hg19_snp138_filtered (перечень не аннотированных в базе данных полиморфизмов, не имеющих rs), chr5.dbsnp.log (отчёт о работе команды).
Из всех найденных мною полиморфизмов из образца 5 хромосомы 24 SNP имеют rs, а 4 - не имеют. Причём последние обладают низким качеством чтений (3.0136 - 21.0411) и плохой глубиной покрытий (1, 3, 8). Все действительно интересующие нас полиморфизмы, расположенные в экзонах, имею аннотацию в базе данных dbSNP.
Аннотация в 1000 Genomes
Версия: 1000g2014oct
Тип аннотации: filter-based annotation (основана на фильтрации)
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr5.1000g -dbtype 1000g2014oct_all
-buildver hg19 chr5.annovar /nfs/srv/databases/annovar/humandb/
В результате я получила файлы chr5.1000g.hg19_ALL.sites.2014_10_dropped (перечень полиморфизмов, имеющих rs в базе данных 1000 Genomes, с указанием частоты их встречаемости), chr5.1000g.hg19_ALL.sites.2014_10_filtered (перечень полиморфизмов, не имеющих rs в базе данных 1000 Genomes), chr5.1000g.log (отчёт о работе команды).
23 полиморфизма оказались аннотированными в базе данных 1000 Genomes, а 5 - неаннотированными. Так как особый интерес для нас представляют полиморфизмы, расположенные в экзонах, то посмотрим на частоты их встречаемости в таблице 4.
| Таблица 4. Частоты встречаемости полиморфизмов в экзонах | ||||||||
| ген | координата (chr5) |
референс | чтение | аминокислотная замена |
тип замены |
глубина покрытия |
качество чтений |
частота встречаемости |
| IL7R | 35861068 | T | С | I66T | гомозиготная | 44 | 221.999 | 0.59984 |
| IL7R | 35871190 | G | А | V138I | гомозиготная | 98 | 221.999 | 0.666933 |
| IL7R | 35874575 | С | Т | T244I | гетерозиготная | 164 | 225.009 | 0.172524 |
| CAPSL | 35910529 | С | Т | R85Q | гомозиготная | 92 | 221.999 | 0.525359 |
Эти данные отлично дополняют комментарий к таблице 3. Мы видим, что у пациента 3 часто встречающиеся мутации не приводят к значимым структурным перестройкам белков, а 1 редко встречающаяся мутация наоборот считается нормой, а не отклонением от нормы.
Аннотация в GWAS (Genome-Wide Association Studies)
Версия: gwasCatalog
Тип аннотации: region-based annotation (основана на разметке других регионов генома)
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out chr5.gwas -dbtype
gwasCatalog chr5.annovar /nfs/srv/databases/annovar/humandb/
В результате я получила файлы chr5.gwas.hg19_gwasCatalog (перечень полиморфизмов, имеющих клиническое значение), chr5.gwas.log (отчёт о работе команды).
5 из 28 полиморфизмов имеют клинические значения, связи с которыми представлены в таблице 5.
| Таблица 5. Связи SNP с клиническими значениями | |||||||
| категория | ген | нукл. замена |
аминокисл. замена |
глубина покрытия |
качество чтений |
частота | клиническое значение |
| exonic | IL7R | С->T | T244I | 164 | 225.009 | 0.172524 | cахарный диабет 1 типа (инсулинозависимый диабет) рассеянный склероз |
| exonic | CAPSL | С->T | R85Q | 92 | 221.999 | 0.525359 | cахарный диабет 1 типа |
| intronic | HMGCR | А->G | нет | 91 | 221.999 | 0.625 | уровень холестерина ЛПНП общий уровень холестерина |
| intronic | HMGCR | С->T | нет | 5 | 58.0073 | 0.405751 | количественные признаки уровень холестерина ЛПНП |
| UTR3 | HMGCR | Т->C | нет | 58 | 225.009 | 0.416134 | уровень холестерина ЛПНП общий уровень холестерина |
Аннотация в ClinVar
Версия: clinvar_20150629
Тип аннотации: filter-based annotation (основана на фильтрации)
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl chr5.annovar /nfs/srv/databases/annovar/humandb/
-filter -dbtype clinvar_20150629 -buildver hg19 -out chr5.clinvar
В результате я получила файлы chr5.clinvar.hg19_clinvar_20150629_dropped (перечень полиморфизмов, аннотированных в Clinvar), chr5.clinvar.hg19_clinvar_20150629_filtered (перечень полиморфизмов, не аннотированных в Clinvar), chr5.clinvar.log (отчёт о работе команды).
Лишь 3 полиморфизма оказались аннотированными в ClinVar. Данные о них подробнее представлены в таблице 6.
| Таблица 6. Связи SNP с заболеваниями | ||||||||
| категория | ген | нукл. замена |
аминок. замена |
глубина покрытия |
качество чтений |
частота | клиническое значение |
возможные заболевания |
| exonic | IL7R | Т->С | I66T | 44 | 221.999 | 0.59984 | патогенна не проверено |
тяжелый комбинированный иммунодефицит |
| exonic | IL7R | G->A | V138I | 98 | 221.999 | 0.666933 | патогенна не проверено |
тяжелый комбинированный иммунодефицит |
| exonic | IL7R | С->T | T244I | 164 | 225.009 | 0.172524 | не проверено | не указано |
Обобщение
Сводную таблицу с описанием всех SNP вы можете скачать по ссылке.