Предсказание генов эукариот
Описание выданного скэффолда
Для работы мне выдали скэффолд NW_003546366, который принадлежит организму Amphimedon queenslandica, который имеет следующее систематическое положение: Eukaryota; Metazoa; Porifera; Demospongiae; Heteroscleromorpha; Haplosclerida; Niphatidae; Amphimedon.

Amphimedon queenslandica (ранее известна как Reniera sp.) - губка, родом из Большого Барьерного рифа. Её фотография представлена выше. Amphimedon queenslandica является модельным организмом различных исследований, посвящённых эволюции развития многоклеточных животных, несмотря на то, что её очень сложно сохранить в неволе. Поэтому её геном полностью секвенирован. Впервые этот организм был обнаружен в 1998 году на рифе у острова Херон (Heron Island Reef) в процессе исследования Салли Лэйс (Sally Leys) различных видов губок. Но формально Amphimedon queenslandica была описана лишь в 2006 году. Как и большинство губок, выданный мне для работы организм имеет двухфазных жизненный цикл. Сначала личинка ведёт планконный образ жизни, а потом вырастает, претерпевает метаморфоз и становится бентосным обитателем. Amphimedon queenslandica являются гермафродитами. Их размножение начнается с выброса сперматозоидов в воду. Затем сперматозоиды с током воды попадают к яйцеклеткам, сидящим внутри особей, и происходит оплодотворение. Эмбрионы развиваются в специальных выводковых камерах, пока они не достигнут определённого размера. После чего они выходят во внешнюю среду и у них начинается личиночная стадия развития.
Ниже в таблице 1 приведена краткая информация о скэффолде NW_003546366.
| Таблица 1. Характеристики скэффолда NW_003546366 | ||||||
| Размер | Биопроект | Биообразец | Сборка | Число генов | Число достоверных генов | Число белок-кодирующих последовательностей (CDS) |
| 231,514 пн | PRJNA66531 | SAMN02743868 | GCF_000090795.1 | 39 | 34 | 40 |
По ссылке вы можете скачать последовательность скэффолда NW_003546366 в FASTA формате.
По данным RefSeq мне удалось обнаружить лишь один ген (LOC100638854) во всей последовательности, для которого предсказывается альтернативный сплайсинг. Ниже представлено его изображение. Стрелки указывают положение гена цепи, в нашем случае ген LOC100638854 находится на прямой цепи. Фиолетовым цветом показаны последовательности мРНК, образующиеся после транскрипции. Области, где фиолетовая линия более тонкая, вырезаются в результате сплайсинга. В итоге остаётся мРНК, состоящая из областей, где фиолетовая линия более толстая. Жирные красные линии указывают на положение белок-кодирующих последовательностей (CDS). Так как в результате сплайсинга вырезаются участки, обозначенные тонкой красной линией, то на одной молекуле мРНК остаётся одна белок-кодирующая последовательность, которая всегда короче мРНК. Дело в том, что по краям CDS располагаются нетранстлируемые области 5'UTR и 3'UTR (соответственно 5' и 3' концы мРНК). 5'UTR, или 5'НТО, содержат такие важные функциональные элементы, как участки внутренней посадки рибосомы (IRES) и внутренние открытые рамки считывания (uORFs). Когда 3'UTR, или 3'НТО, могут содержать сайты связывания с микроРНК, которая блокирует трансляцию мРНК и вызывает её разрушение, или содержать сигналы полиаденилирования, отвечающие за присоединение к 3'-концу мРНК поли-А хвоста. Поли-А хвост играет важную роль в транспорте мРНК из ядра, её трансляции и стабильности. Со временем он укорачивается, и, когда его длина станет достаточно малой, мРНК разрушается под действием специальных ферментов. Ещё 5'-конец мРНК соединён с кэпом (Cap), который участвует в регуляции транскрипции, сплайсинга, процессинга, транспорта, защиты от эндонуклеаз, трансляции мРНК.

Мы видим из изображения, что данному гену соответствуют две изоформы X1 (XP_011404093.1) и X2 (XP_003386827.1), верхняя и нижняя соответственно. Однако обе они недостоверны и предсказаны программой Gnomon. Хотя авторы статьи утверждают, что в данных мРНК есть сходства с разными EST (короткими последовательностями ДНК, которые используют для определения последовательностей генов) и с некоторыми уже аннотированными последовательностями РНК.
Предсказание генов и белок-кодирующих областей с помощью web-сервера AUGUSTUS
Сперва я перешла на web-сервер AUGUSTUS. В качестве входных параметров я указала организм Amphimedon queenslandica (animal), из генома которого будут браться параметры модели, и файл scaffold.fasta. Остальные параметры я оставила по умолчанию. Ниже представлено изображение параметров для запуска программы, которые я использовала.
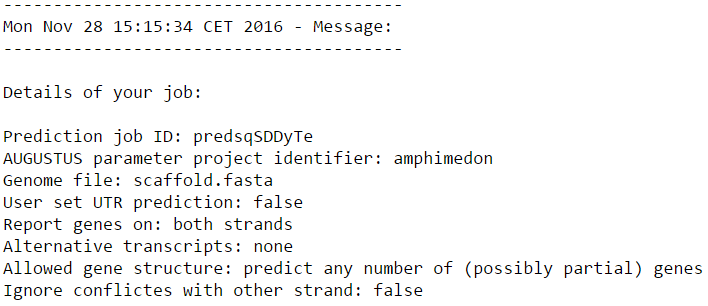
- Prediction job ID - идентификатор моего запроса о предсказании генов.
- AUGUSTUS paremeter project identifier - указание организма, из генома которого будут браться параметры модели для работы программы. Можно загрузить геном в виде архива, ввести идентификатор проекта по сборке генома или указать организм из предложенного списка. Это поле обязательно для заполнения.
- Genome file - вводится файл с одной или несколькими последовательностями ДНК, для которых я хочу предсказать гены. Вместо файла можно также ввести интернет ссылку на файл с геномом. Это поле обязательно для заполнения.
- User set UTR prediction - можно попросить программу также предсказывать области UTR, тогда напротив этого поля будет написано "true". По умолчанию стоит "false".
- Report genes on - программа может искать гены на обеих цепях ДНК (both strands), только на прямой цепи (forward strand only) или только на комплементарной цепи (reverse strand only). По умолчанию выбраны обе цепи.
- Alternative transcripts - программа может на выходе давать разное количество возможных альтернативных транскриптов (few, medium, many) или одному гену сопоставлять один транскрипт (none). Так как в моём скэффолде был всего один ген, кодирующий две изоморфы, то для ускорения работы программы я выбрала "none". По умолчанию стоит "none".
- Allowed gene structure - программа может предсказать любое число (возможно частичных) генов (predict any number of (possibly partial) genes), так как во входной последовательности гены могут быть представлены не полностью, например, иметь чёткое начало, а конец в неотсеквенированной области (много букв N). Эта опция выбрана по умолчанию. Можно попросить программу предсказывать только целые гены (predict only complete genes) или по крайней мере один целый ген, если это возможно (predict only complete genes - at least one). Также программа может предсказать только один целый ген (predict exactly one complete gene).
- Ignore conflictes with other strand - по умолчанию считается, что гены даже на противоположных цепях не перекрываются (false). Но программа может предсказывать гены на каждой цепи отдельно и независимо (true), что в некоторых случаях необходимо, хотя в большинсве своём приводи к ошибочным предсказаниям.
В результате работы программы я получила архив predictions.tar.gz, содержащий 6 файлов: augustus.aa, augustus.cdsexons, augustus.codingseq, augustus.gbrowse, augustus.gff, augustus.gtf. В таблице 2 представлено описание содержания каждого файла. В них g1, g2, ..., g60 - номера предсказанных генов; t1 - номер транскрипта; cds1, cds2, ..., cds38 - номера белок-кодирующих последовательностей.
| Таблица 2. Описание содержания файлов, выдаваемых программой AUGUSTUS | |
| Файл | Содержимое |
| augustus.aa | Последовательности белков в формате FASTA для каждого из предсказанных генов и транскриптов. По 100 аминокислотных остатков на одной строчке файла. |
| augustus.cdsexons | Белок-кодирующие последовательности (CDS) после сплайсинга (экзоны) в формате FASTA для каждого предсказанного гена и транскрипта. Одна последовательность CDS на одной строчке файла. Нумерация cds ведётся в соответствии с их расположением относительно прямой цепи, т.е. для гена из обратной цепи участок cds1 будет последним и содержащим стоп-кодон. |
| augustus.codingseq | Кодирующие последовательности предсказанных генов (без интронов) в формате FASTA с учётом цепи, то есть начало записи последоватальности в файле совпадает с 5'-концом кодирующей цепи. Первая строчка записи содержит 79 пн, последующие - по 98 пн. |
| augustus.gff | В начале файла представлена аннотация к работе программы, затем для каждого предсказанного гена идёт таблица, в которой указаны: название элемента (ген, транскрипт, старт-кодон, терминальные и внутренние участки, интроны, участки CDS и стоп-кодон), координаты начала и конца относительно прямой цепи ДНК, направление цепи, кодирующей предсказанный ген, и идентификатор последовательности. После каждой таблицы приведена кодирующая последовательность гена и предполагаемая последовательность белка. |
| augustus.gtf | Как в файле augustus.gff, но только таблица не содержит строчки, помеченные символом #. |
| augustus.gbrowse | Та же таблица, как в файле augustus.gtf, только слово "transcript" заменено на "mRNA" и компактнее записаны идентификаторы. |
Для дальнейшей работы возьмём файл с предсказаниями генов augustus.gbrowse. С помощью программы gbrowse.py я получила файл gbrowse.txt. Со страницы NW_003546366 я скачала файл scaffold.gb, а затем с помощью программы featcopy пакета EMBOSS я получила файл scaffold.gff, содержащий таблицу аннотаций особенностей.
featcopy scaffold.gb scaffold.gff
Далее с помощью программы gff2.py я из файла scaffold.gff получила файл scaffold.txt. Теперь можно приступить к непосредственному сравнению предсказания программы AUGUSTUS с генами, указанными в скэффолде. Для этого я воспользовалась написанной ранее программой comparison.py. На вход я дала файлы scaffold.txt и gbrowse.txt.Ниже представлен результат её работы.
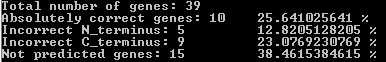
Всего программа AUGUSTUS предсказала 60 генов. Из них 10 совпали полностью, 5 имели ошибочный 5'-конец (N-конец), 9 имели ошибочный 3'-конец (С-конец), а остальные не имели совпадений. В результате 15 из 39 генов, указанных в скэффолде, не были предсказаны ни по одному концу.