Нуклеотидный BLAST
Нахождение гена кодирующего δ-субъединицу АТФ-синтазы
В файле, полученном при выполнении предыдущего практикума, с последовательностями белков был найден (с помощью текстового поиска) белок аннотированный как ATP synthase subunit delta. Было найдено два совпадения: XP_073700398.1 и XP_073700399.1 (дальнейшая работа велась с XP_073700398.1). Последовательность этого белка была сохранена в отдельный файл, для дальнейшей работы с ним.
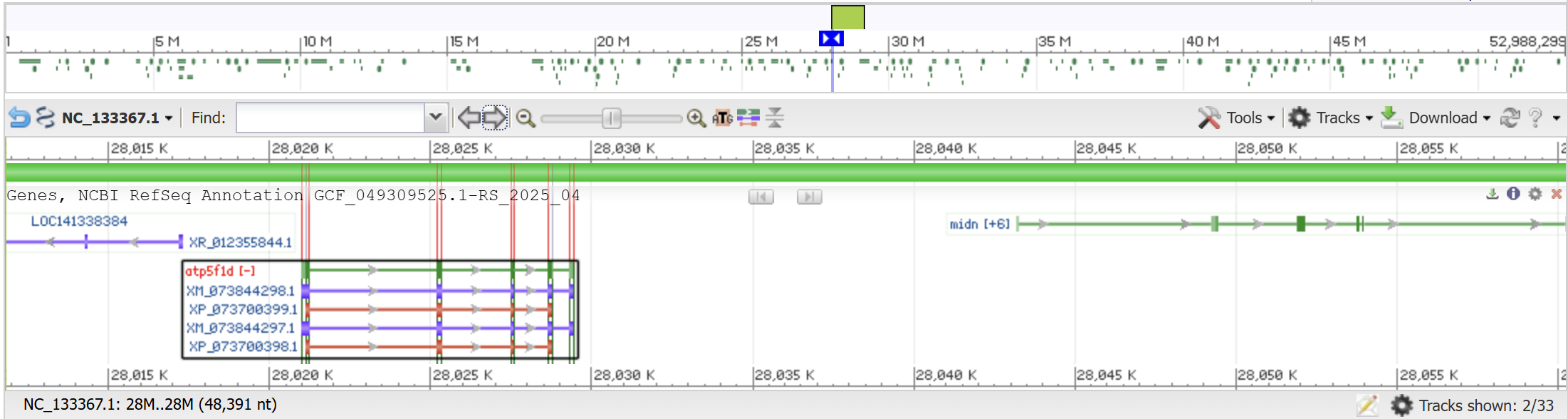
Затем в файле с подробной информацией о генах моего организма (gbff), я нашёл необходимый белок по текстовому поиску. Затем нашёл локус в котором он находится, а именно: NC_133367.
Далее я нашёл по данному идентификатору соответсвующую запись в NCBI Nucleotide и нашёл расположение моего белка на этом участке (это 7 хромосома). Скачал последовательность участка генома, который кодирует нужный белок (ссылка на файл). Ниже приведён учаток (рис. 1), на котором расположен ген.
Варианты BLAST для фрагмента ДНК
Выбранный мной эукариот относится к вторичноротым, поэтому для это задания, как отдолённое семейство первичноротых было выбрано Araneae (пауки).
С помощью алгоритма blastn, с изменённой длиной слова (word size = 7) и Expect threshold = 1e-9, по базе данных refseq_genomes был произведён поиск похожих нуклеотидных последовательностей. Был выбран blastn (а не megablast), так как поиск производится по отдолённому семейству, а megablast лучше находит почти идентичные последовательности. Также была уменьшена длина слова с 11 до 7 и Expect threshold с 0,05 до 1e-9, чтобы было больше находок, но они были специфическими.
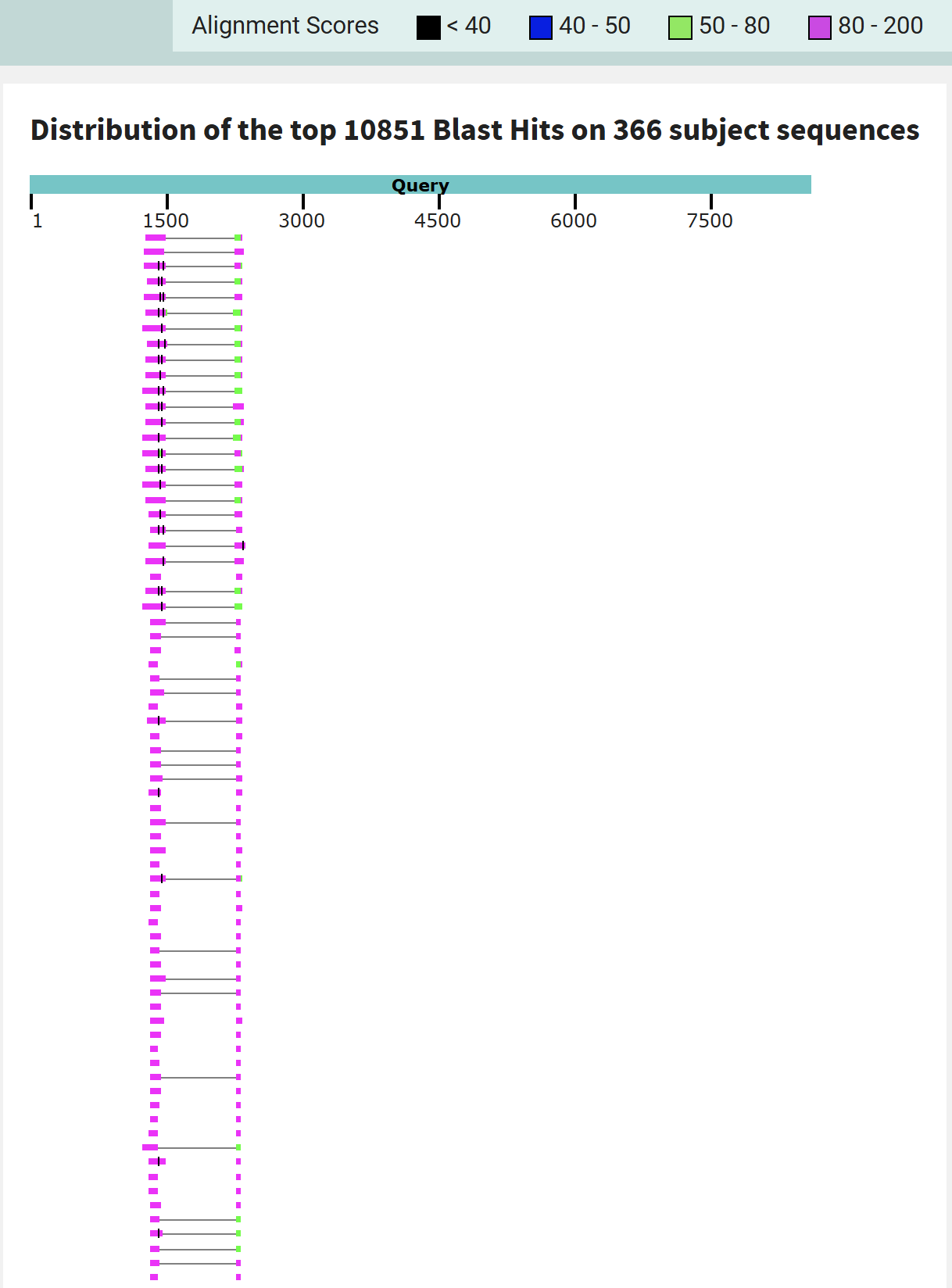
Выдача оказалась очень странной. Если менять Expect threshold на 0,005, то находок будет 995, что очень много. Если понижать этои порог до 1e-9, то находок станет меньше 366, но это всё ещё очень много. Более того, примечательно, что совпавшие участки имеют хороший вес (рис. 2).
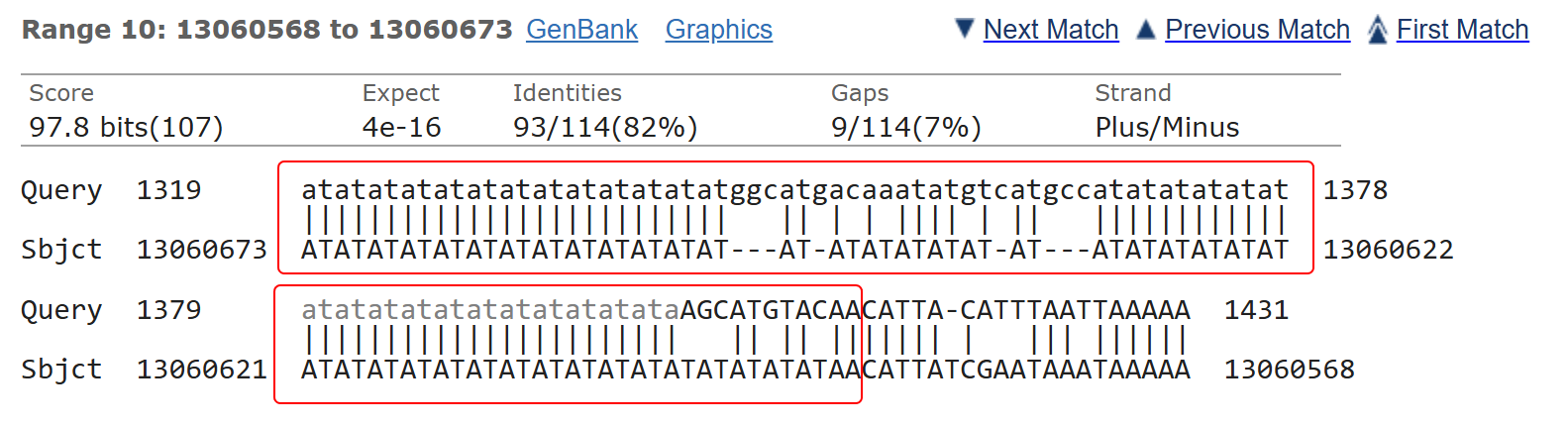
Однако посмотрев на выравнивания, можно заметить, что в большом количестве случаев была выравнена повторяющаяся последовательность, а именно ATATATA..ATATAT (один из примеров рис. 3). Если посмотреть на координаты гена, по которым произошло такое выравнивание, то можно увидеть, что данный участок явдяется интроном. Этим можно объяснить такое огромное число находок.
Затем был применён алгоритм tblastn к последовательности белка, чтобы также найти гомологи в геномах пауков. База данных - refseq_genomes, семейство - Araneae (пауки). Также можно было использовать алгоритм tblastx, но он медленее. Длина слова была установлена на 2, для большего числа находок.
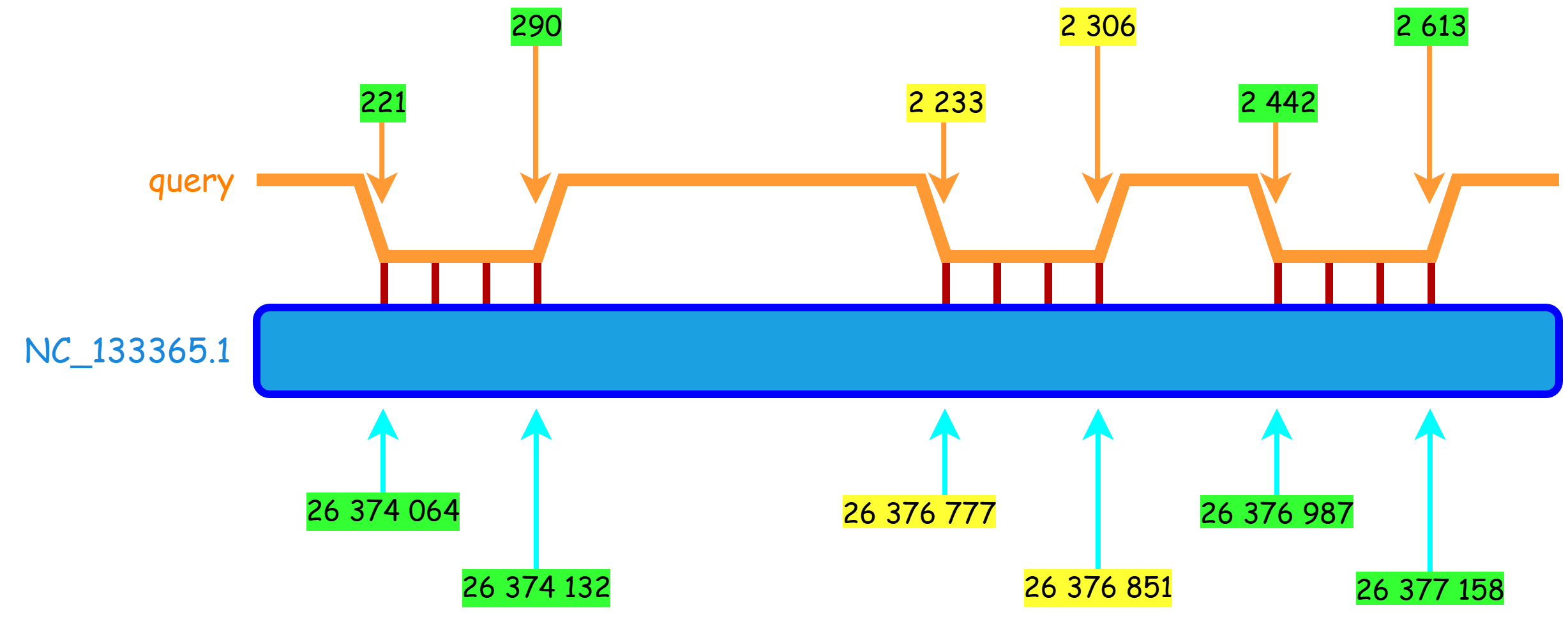
Результаты полученные данным методом более ожидаемые, но они сильно отличаются от выдачи blastn. Находки 4, все они встречались в выдаче blastn. На рис. 4 представлен графический результат поиска.
Нахождение в геноме эукариота гены основных рибосомальных РНК по далекому гомологу
С помощью следующей команды была построена база данных для дальнейшего применения blast на локальном компьютере:
makeblastdb -in .\GCF_049309525.1_GarRuf1.0_genomic.fna -dbtype nucl
Далее был применён алгоритм blastn для последовательностей 16S и 23S рРНК E. coli по новой базе данных. Был применён именно blastn, так как последовательности рРНК консервативны и их можно найти даже у отдалённых организмов. Ниже приведены команды, с помощью которых это было сделано:
blastn -task blastn -query .\16S_rRNA_ecoli.fasta -db .\GCF_049309525.1_GarRuf1.0_genomic.fna -outfmt 7 -evalue 0.05 -out res_16S
blastn -task blastn -query .\23S_rRNA_ecoli.fasta -db .\GCF_049309525.1_GarRuf1.0_genomic.fna -outfmt 7 -evalue 0.05 -out res_23S
Ссылки на результаты для 16S рРНК и 23S рРНК.
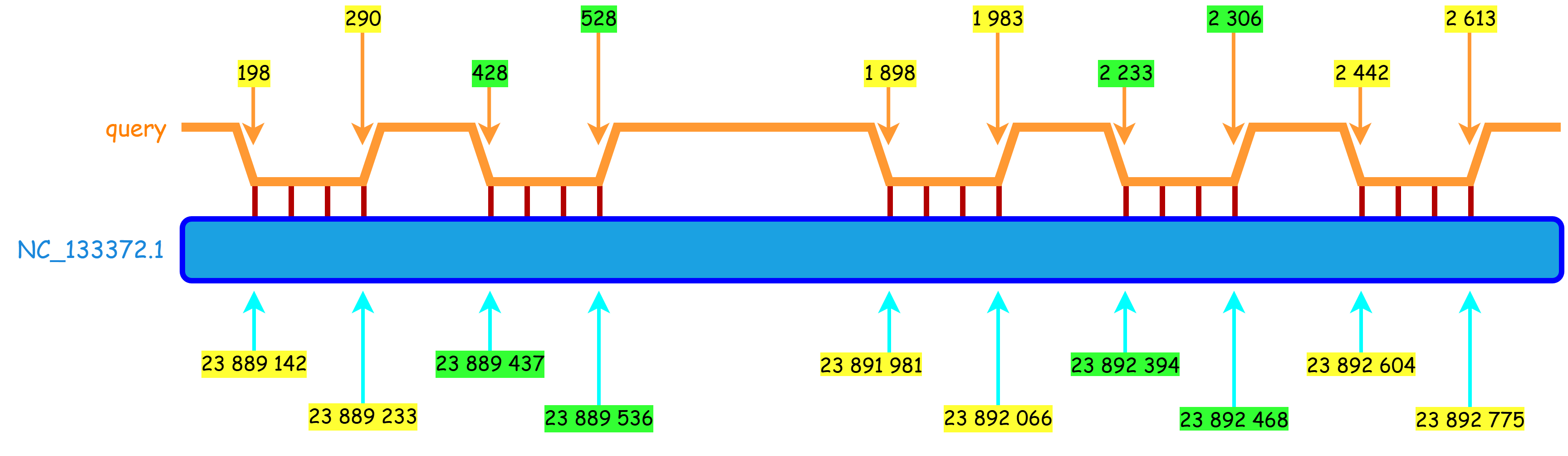
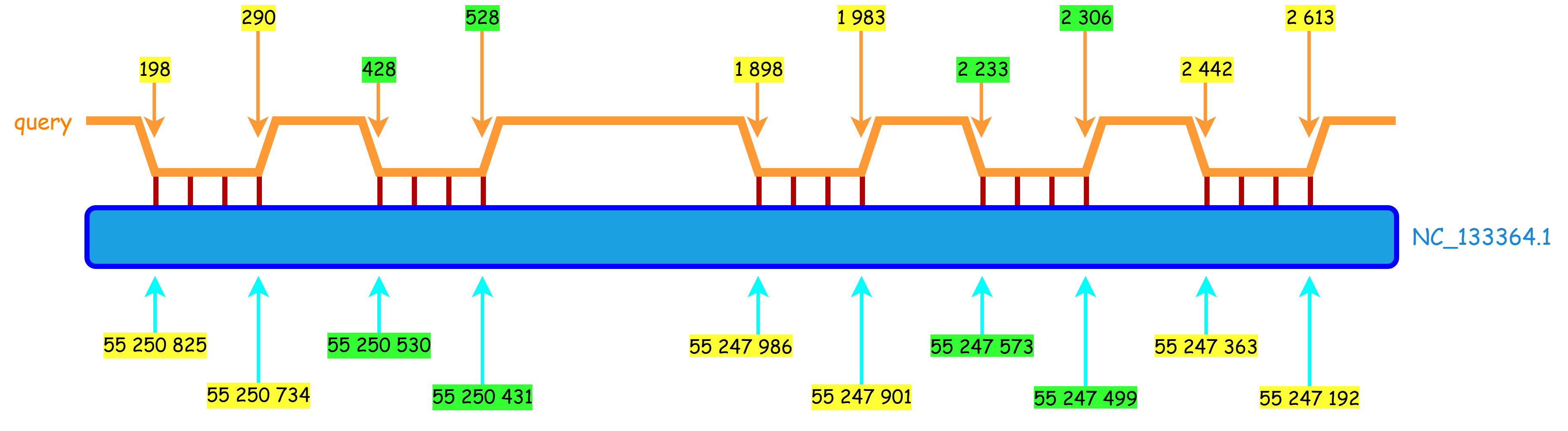
Также на рисунках 5, 6, 7 представлены схемы находок для 23S рРНК алгоритма бласт (только те, которые не являются одной строчкой выдачи программы).
Подберите пару геномов и постройте карты их локального сходства
Для этого задания необходимо было найти бактерий для которых, выдача программ blastn и megablast заметно отличается. Также лимитирующим фактором для выбора была длина генома, так как помимо упомянутых программ надо также использовать tblastx, на вход которому не всегда получается давать большие последовательности (больше 1 Mb уже не всегда работает).
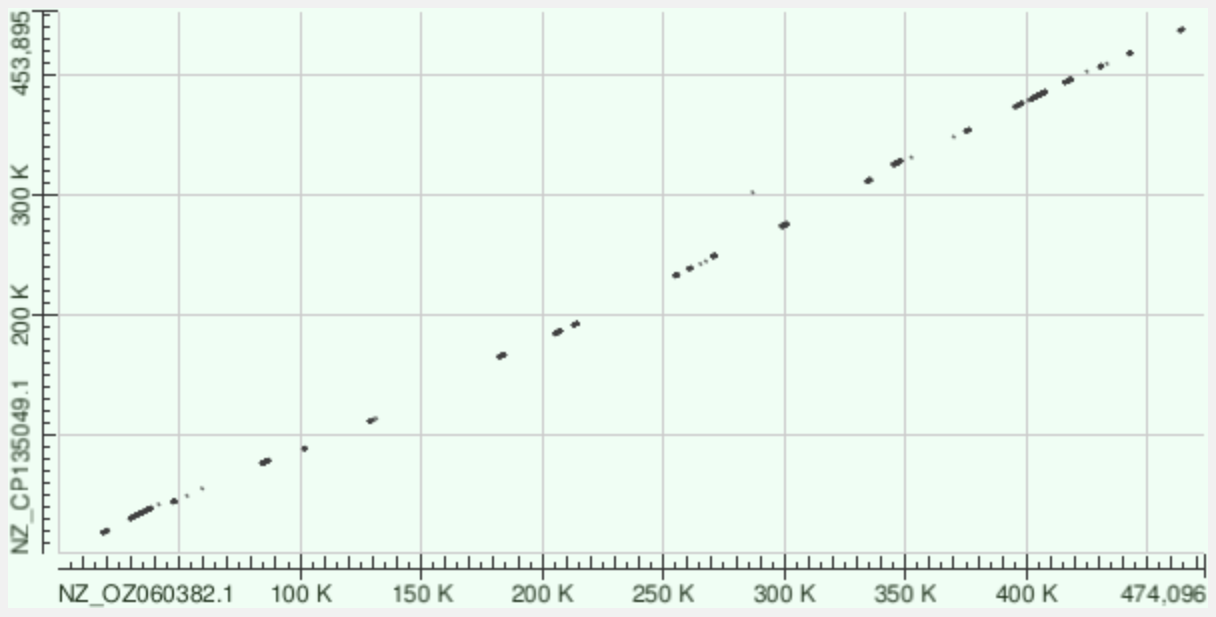
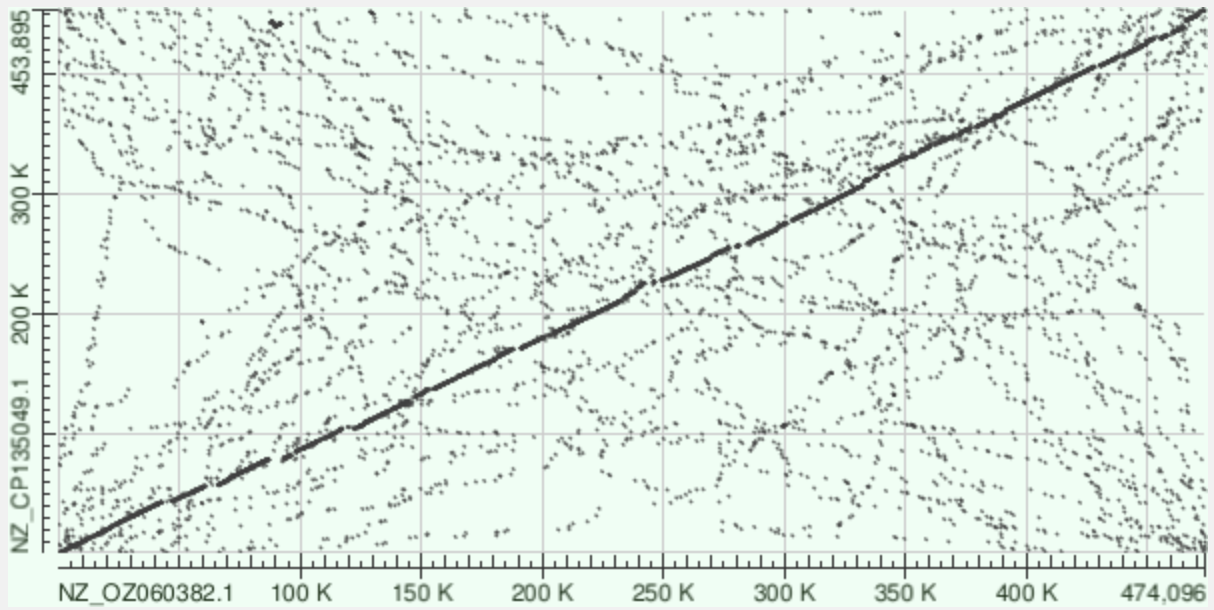
Мною были выбраны бактерии: Buchnera aphidicola (Chaitophorus populicola) (NZ_OZ060382.1) и Buchnera aphidicola (Taiwanaphis decaspermi) (NZ_CP135049.1). Искал я среди бактерий, уровень сборки которых комплит. Всех бактерий отсортировал по длине генома.
Далее к геномам были применены программы blast для построения карт локального сходства (рис. 8, 9, 10).