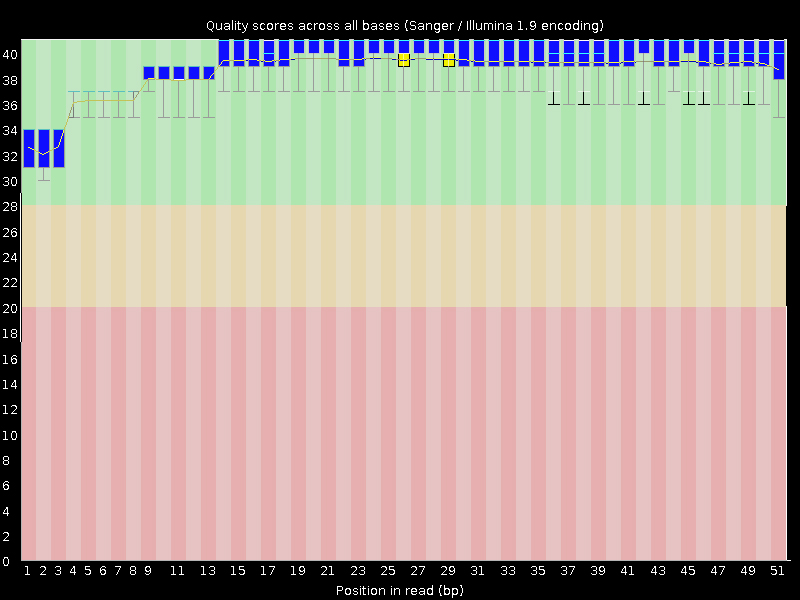
Исходно было получено 24156 чтений. С помощью Trimmomatic с конца каждого чтения были отрезаны нуклеотиды с качеством ниже 20, после чего оставлены только чтения длиной не меньше 50 нуклеотидов. После очистки осталось 24051 ридов. Разница в качестве чтений до и после очистки отображена на Рис. 1. Видно, что различия есть не везде и в целом довольно незначительные.
Картирование ридов
23563 чтений были картированы на хромосому. При этом параметр --no-spliced-alignment не использовался, поскольку в случае рнк вполне логично ожидать сплайсинга, поэтому целесообразно разделять транскрипт на фрагменты при необходимости.
Подсчет чтений
Для подсчета чтений была испольована программа htseq-count, имеющая в т.ч. следующие параметры:
-f: формат входного файла.
-s: к какой цепи относятся риды.
-i: feature ID.
-m: действия с ридами, попадающими на несколько генов.
Результаты подсчета
Обработано 24061 SAM выравнивание.
ENSG00000181163.9 22438 ENSG00000249353.2 314 __no_feature 791 __not_aligned 498 __alignment_not_unique 20
T.e. 22438 чтений приходятся на ген ENSG00000181163.9, 314 - на ENSG00000249353.2, 791 - ни на какой ген, 20 выравниваются неоднозначно.
Оба найденных гена свзяны с нуклеофозмином - ядрышкомым белком, отвечающим за работу хроматина, который в т.ч. способствует правильной сборке гистонов, поддерживает геномную стабильность, предотвращает апоптоз при локализации в ядрышке и т.д.
Использованные команды:
| fastqc chr5.1.fastq | Проверка качества чтений |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr5.1.fastq chr5.1_trimmed.fastq TRAILING:20 MINLEN:50 | Очистка чтений |
| fastqc chr5.1_trimmed.fastq | Проверка качества после очистки чтений |
| hisat2 -x chr5 -U chr5.1_trimmed.fastq -S chr5.1_aligned.sam --no-softclip | Выравнивание прочтений и референса |
| samtools view -b chr5.1_aligned.sam -o chr5.1_aligned.bam | Перевод выравнивания в бинарный формат |
| samtools sort chr5.1_aligned.bam chr5.1_aligned_sorted | Сортировка выравнивания по координате в референсе |
| samtools index chr5.1_aligned_sorted.bam | Индексирование отсортированного файла |
| htseq-count -f bam chr5_aligned.bam -i gene_id -s no gencode.v19.chr_patch_hapl_scaff.annotation.gtf > count.txt | Подсчет чтений |
| grep -wv 0 count.txt > count_not_empty.txt | Создание файла с числом чтений только для генов, на которые чтения в принципе были картированы. |