Анализ качества и очистка чтений
Дан файл с чтениями генома резуховидки Arabidopsis thaliana. Сначала проверим их качество с помощью сервиса FastQC. Ее отчет доступен по ссылке и на рисунке 2.
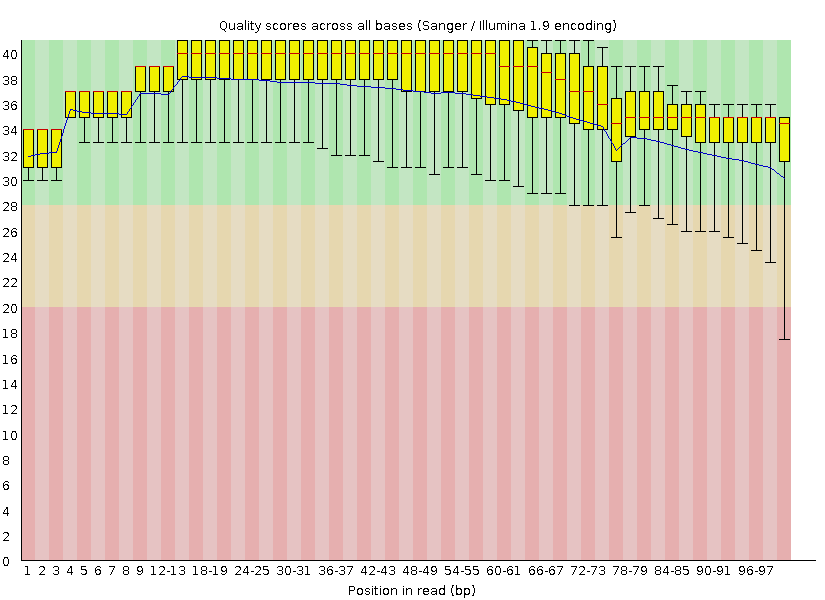
Далее была проведена очистка (тримминг) чтений с помощью программы Trimmomatic. Были удалены адаптеры, нуклеотиды с плохим качеством (<20) с конца каждого прочтения, и оставлены только прочтения длины не менее 50. Затем опять сделали анализ качестыва с помощью FastQC. Команда для запуска Trimmomatic:
fastqc read.gz
java -jar /usr/share/java/trimmomatic.jar SE read.gz out.fastq ILLUMINACLIP:adapters.fasta:2:7:7
java -jar /usr/share/java/trimmomatic.jar SE out.fastq out.fastq TRAILING:20
java -jar /usr/share/java/trimmomatic.jar SE out.fastq out.fastq MINLEN:50
fastqc out.fastq
Был выбран формат phred33, так как использован метод секвенирования Illumina 1.8. Количество ридов сократилось с 4 млн до 10 тысяч. Отчет FastQC обработанных чтений доступен по ссылке и на рисунке 2.
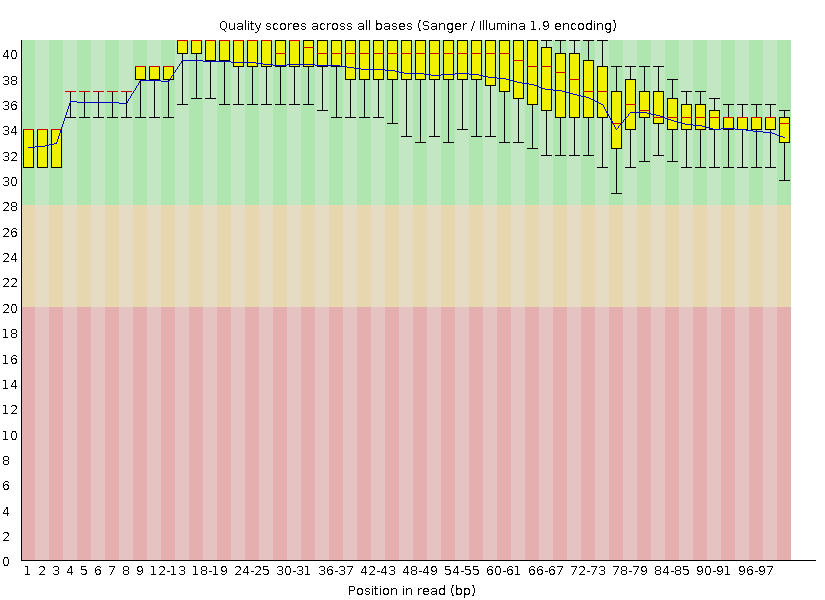
Среднее качество последнего нуклеотида сильно выросло, это видно на графике Per base sequence quality. На графике частоты встречаемости букв на каждой позиции (Per base sequence content) соотношение нуклеотидов остается таким же, как и до очистки: аденина+тимина примерно в два раза больше, чем гуанина+цитозина. Однако график о ридах до очистки более сглажен, чем второй. Это связано с уменьшением количества длинных чтений, то есть с увеличеснием статистической погрешности вычислений.