Нуклеотидный BLAST
Задание 1.Определение функции и таксономии нуклеотидной последовательности
В рамках первого задания нужно было предположить функцию нуклеотидной последовательности, полученной в ходе расшифровки хроматограммы, и таксономическое положение организма, из клеток которого она была выделена. В данном случае уместно использовать алгоритм blastn, так как мы не знаем точно, имеется ли точно такая последовательность в базе данных (megablast может не дать результата) и ищем сколько-нибудь близкие. В качестве query sequence взят контиг, полученный в ходе практикума ранее. База данных - Nucleotide colection (nr/nt), так как неизвестно, есть ли интересующая нас последовательность среди аннотированных. Значение max target sequences оставлено по умолчанию равным 100, expect threshold - 0.05, размер слова word size задан минимальный возможный (7) для повышения чувствительности поиска. Значения scoring parameters также оставлены по умолчанию
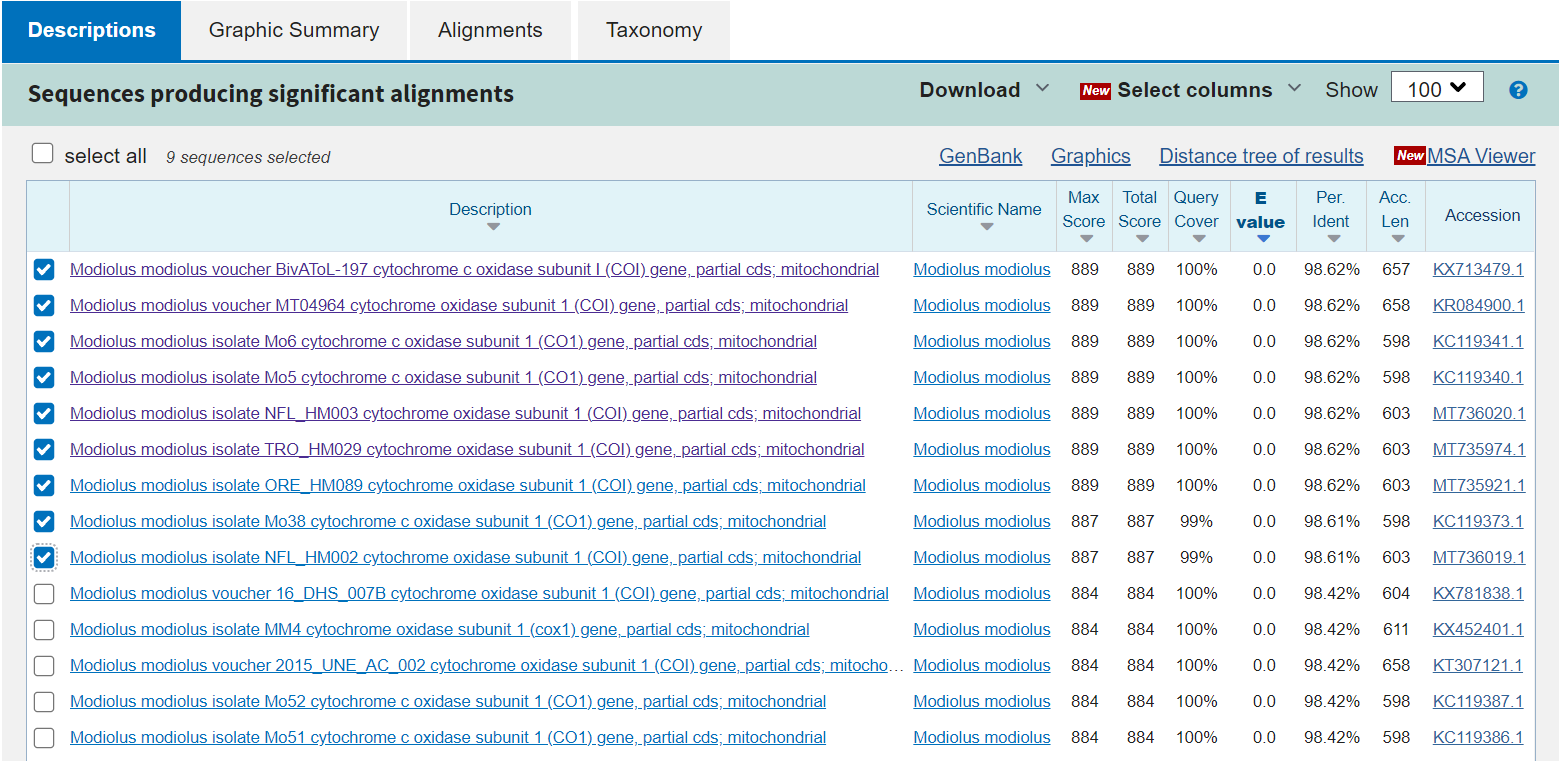
Для всех 100 полученных в результате находок величина покрытия составила 99-100%, значение e-value - машинный ноль, очень мало, а процент совпадения - от 89.53% до 98.62%. Все находки принадлежат Modiolus modiolus - двустворчатому моллюску, также известному как конская мидия. Согласно традиционно используемому источнику, встречается вдоль атлантического и тихоокеанского побережья Северной Америки практически во всех широтах, а также в северной части атлантического побережья Евразии, наиболее распространены в Шотландии. Также все находки представляют собой гены 1 субъединицы белка цитохром-c-оксидазы - одного из ферментов аэробной дыхательной цепи. Вполне ожидаемо, что этот ген митохондриальный.

В силу того, что все 100 находок достаточно хорошо совпадают с интересующей нас последовательностью, можно утверждать, что она является, вероятнее всего, митохондриальным геном конской мидии Modiolus modiolus, кодирующим цитохром-с-оксидазу. Далее представлены ссылки на выдачу BLAST, выравнивание для первых 9 лучших находок в текстовом виде и в формате fasta и проект Jalview. Изначально для выравнивания в Jalview были также выбраны первые 9 находок, но выравнивание явно разбилось на 6 находок, практически полностью совпадающих с искомой последовательностью, и три выравнявшихся значительно хуже. Это объясняется тем, что для части генов на query выравнивается прямая цепь (1-506 нуклеотиды искомой последовательности соответствует 87-592 в последовательности из базы), а для других - обратная (539-34 нуклеотиды последовательности из базы). Это видно в выравнивании, которое выдает BLAST. Очевидно, выравнивать их одновременно не имеет смысла, поэтому в конечном представленном здесь варианте проекта представлены последовательности, найденные для прямых цепей. Ко всему прочему, результат выполнения данного задания позволяет отредактировать контиг в тех местах, где было решено оставить "двусмысленные" коды нуклеотидов. Вероятнее всего, на 3-й позиции (оставлено K - G или T) находится гуанин, на 15-й (оставлено R - A или G) - аденин, а на 70-й (оставлено S - C или G) - цитозин. Разумеется, существует вероятность мутаций и в выравнивании рассматривается очень мало последовательностей, что не позволяет утверждать точно, но предположение сделать реально.
Задание 2.Поиск генов белков в неаннотированной нуклеотидной последовательности
Целью следующего задания было предположить наличие гена и определить функцию закодированного в нем белка, используя самстоятельно выбранный контиг или скэффолд. В качестве организма я выбрала Saccharomyces cerevisiae, так как это очень хорошо изученный организм, и, вероятнее всего, в контиге найдутся гены. Код полногеномной сборки на сайте NCBI - WMJW01000042.1, длина контига - 81821 пара нуклеотидов. Для поиска белков был выбран алгоритм blastx, при этом база данных ограничена аннотированными белками (UniProtKB/Swiss-Prot(swissprot) ), чтобы можно было с большей вероятностью доверять полученным данным, а параметр Organism настроен так, чтобы из результатов исключались гены, принадлежащие Saccharomyces cerevisiae. В дополнительных настройках значение Max target sequences оставлено по умолчанию 100, Expect threshold изменено на 10 для повышения чувствительности, значение длины слова word size изменено на 3 с той же целью. Остальные параметры оставлены по умолчанию.
В результате получена следующая выдача BLAST. С достаточно большой вероятностью можно предполагать, что в выбранном контиге действительно содержатся гены белков. Для первых 8 находок значение e-value представляет собой машинный ноль, для остальных очень мало. Процент покрытия около 1%, что неудивительно, если вспомнить размер контига. Есть находки сочень большим процентов идентичности (94.46%, 82.69%). На изображении, представленном слева, показаны все найденные идентичные участки, и по нему видно, что можно предположить наличие двух генов. Первый от начала последовательности участок - возможный ген митохондриальной цитрат-синтазы, он находится в пределах 11063-9726 нуклеотидов (обратная цепь), второй - ген фосфоглицерат киназы, он расположен на прямой цепи в пределах 26200-27444 нуклеотидов. В прикрепленном текстовом файле с выдачей содержатся 16 находок: с идентичностью выше 68% для фосфоглицерат киназы и выше 66% для цитрат синтазы. Если ориентироваться на процент совпадения, можно сказать, что с наибольшей вероятностью в контиге закодирован ген фосфоглицерат киназы.
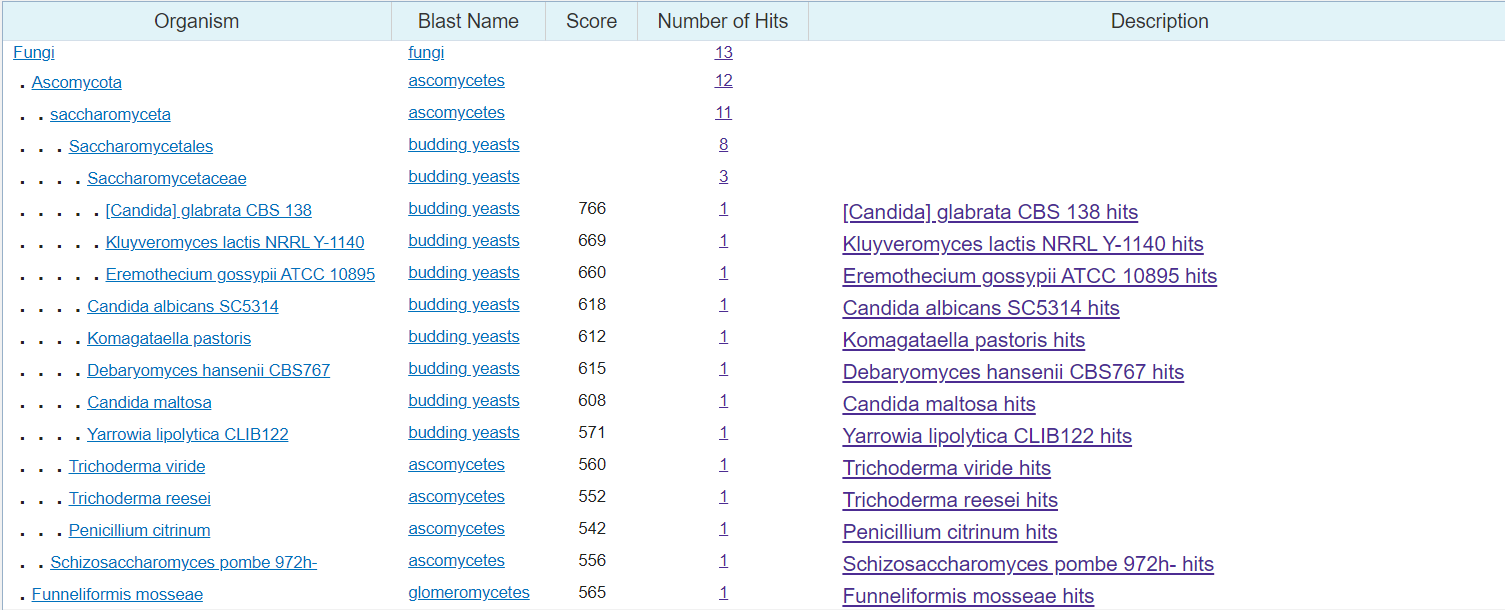
Далее представлена схема, отражающая таксономическое положение организмов, у которых похожий участок кодирует фосфоглицерат киназу. Как можно видеть, все это грибы и большинство - аскомицеты, но запрос сделан для последовательностей, совпадающих с запросом не менее чем на 70%. Если выбрать все 100 находок, в списке организмов будут в том числе Mus musculus, Gallus gallus, Drosophila melanogaster, Arabidopsis thaliana, Clostridium tetani - что свидетельствует о распространенности гена и важности белка в метаболизме. Фосфоглицерат киназа является одним из ферментов гликолиза и катализирует реакцию образования АТФ.
Задание 3. Интепретация карты локального сходства гомологичных хромосом двух бактерий.
Для построения карты локального сходства были выбраны две бактерии рода Yersinia: Yersinia canariae(сборка NZ_CP043727.1) и Yersinia entomophaga(сборка NZ_CP010029.1). Для обеих бактерий в сборке содержалось по одной хромосоме, fasta-файлы доступны далее по ссылкам: Y.canariae и Y.entomophaga. В качестве алгоритма выравнивания был выбран megablast, так как цель работы - сравнить близкие последовательности. При формировании запроса в качестве query sequence была выбрана Y.canariae, subject sequence - Y.entomophaga. Все параметры были оставлены по умолчанию, длина слова для алгоритма megablast составляет 28.
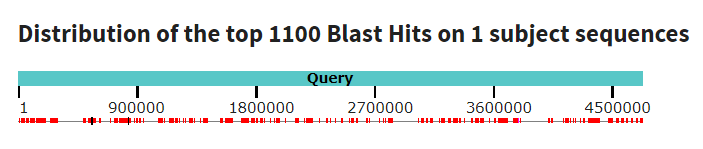
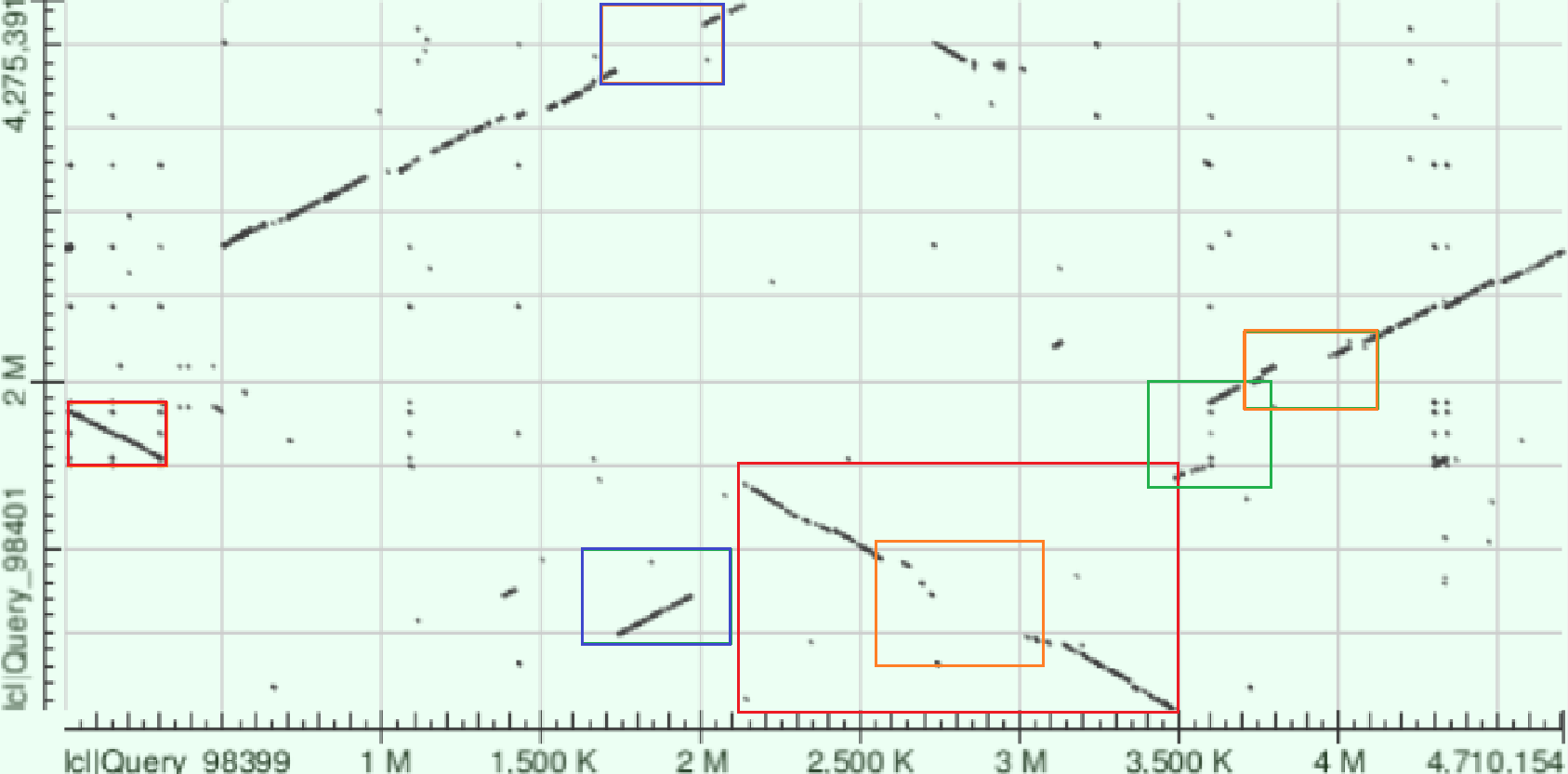
В результате была получена следующая выдача BLAST. Анализируя полученные данные, можно сказать, что хромосомы данных бактерий достаточно различаются: процент идентичности составляет 85.36%, при этом покрытие всего 40%. Но по графику Dot Plot, который представлен выше, видно, что имеются продолжительные участки сходства.
На полученной карте есть несколько участков, требующих объяснения. Вместо одной диагональной линии мы видим две смещенные относительно центра, что может быть связано с ошибкой при расшифровке кольцевого бактериального генома и поиске ориджина репликации. Красными прямоугольниками выделены инверсии - мутации, представляющие собой поворот участка ДНК на 180 градусов. В случае меньшего из двух инвертированных участков можно предположить также наличие инделя, так как если мысленно инвертировать его, он не соединится с основной линией. Зеленым прямоугольником выделен индель - вероятно, участок, расположенный после 3600-тысячного нуклеотида, был вставлен в геном. Также в этом участке произошла делеция, выделенная оранжевым цветом. Внутри длинного инвертированного участка есть неконсервативная область, наличие шума указывает на точечные совпадения. На промежутке 1750-2000 тысяч нуклеотидов, вероятно, наблюдается транслокация, смещенные относительно друг друга участки также выделены на рисунке синим цветом.