Домены и профили
Задание 1. Построение и проверка работы НММ-профиля семейства белков.
Целью данного задания было выявление критерия отнесения конкретного белка к семейству с определенной доменной архитектурой. В качестве исследуемого семейства я выбрала белки, содержащие домен STN1_2 (PF09170) из базы данных Pfam. Домен приблизительно удовлетворяет указанным критериям: в full 467 последовательностей (сравнительно немного), средняя длина 149.9, сходство 48%, покрытие домена - 43.37% (достаточно, чтобы предположить, что в последовательности могут быть еще домены). В разделе Domain organization 11 вариантов доменной архитектуры, из которых два подходят под критерии: включают два домена, содержатся в более чем 20, но менее чем в половине последовательностей, - 207 последовательностей, включающих расположенные рядом домен STN1_2 и домен антикодоновой цепи тРНК, и 112 последовательностей, включающих STN1_2 и домен регулятора теломер Stn1. В обоих случаях домен STN1_2 в структуре расположен вторым. Для исследования была взята вторая архитектура из упомянутых, её схему можно видеть на схеме ниже. Помимо упомянутых 112 белков, подобную архитектуру имеют еще 4, отличие которых состоит в том, что домен STN1_2 в их последовательности прерывается неконсервативной областью.
Число белков с данным доменом в выборке Seed, Full и Uniprot составляет 22, 467 и 803 соответственно, эти данные доступны со страницы Alignments. Длина профиля НММ из базы данных - 167.

Кратко о функции, связанной с доменом: STN1 является компонентом комплекса CST, который связывается с одноцепочечной ДНК и предохраняет теломеры от расщепления ферментами. Характерной чертой комплекса является неспецифичное к последовательности связывание с ДНК. Также рассматриваемый белок содержит структурные мотивы, обеспечивающие возможность взаимодействия с различными другими белками, которые действуют на теломеры.
Для выбранного семейства был скачан fasta-файл с полными последовательностями всех белков. В процессе работы оказалось, что он содержит последовательности с повторяющимися идентфикаторами, поэтому для дальнейшей работы, в частности, с HMM-профилем, был написан скрипт и создан файл, содержащий только уникальные последовательности. Вероятно, это одна из особенностей базы данных, в которых я пока что не разобралась. Уникальных последовательностей среди скачанных 467 оказалось 441. Для создания выборки белков с рассматриваемой доменной архитектурой был скопирован список идентификаторов, после чего посредством скрипта нужные последовательности (причем только уникальные) были собраны в новый файл. Далее с помощью программы MUSCLE было получено их выравнивание. Дальнейшая его обработка осуществлялась в программе Jalview (ссылка на проект).
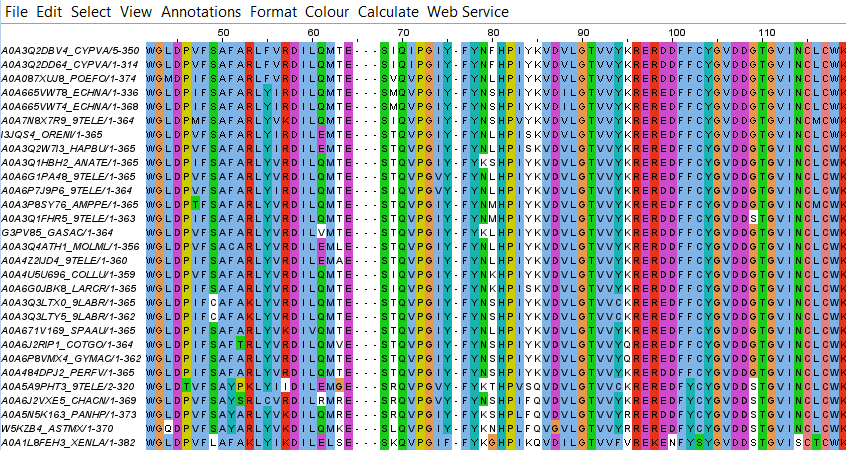
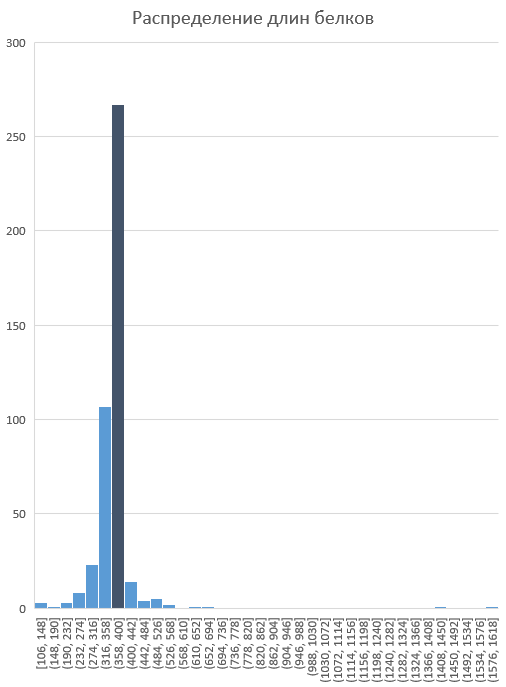
Согласно данным Pfam, координаты домена Snt1 - 22:147, домена STN1_2 - 209:377. Исходя из этого, а также из последовательностей выборки seed рассматриваемого домена в разных белков, которые доступны во вкладке Alignments - Format an alignment, были обрезаны участки выравнивания перед первым и после второго домена. Несколько белков, последовательности которых имели слишком много несоответствий в консервативных для семейства регионах, были удалены. Также были удалены последовательности, имеющие 99-100 процентов сходства. В итоге было получено новое выравнивание, которое уже использовалось для построения HMM-профиля. Данные о белках семейства, вхождении в из последовательность второго рассматриваемого домена, включении их в выборку для построения профиля и длине последовательности отражены на листе sample таблицы. На том же листе таблицы представлена гистограмма длин белков, из которой видно, что подавляющее число белков семейства имеет длину в пределах 358-400. Приблизительно такую длину имеют и вырезанные участки с двухдоменной архитектурой. Длины последовательностей также были посчитаны с использованием файла с уникальными последовательностями и возможностей языка Python.
Для построения HMM-профиля были последовательно выполнены следующие команды:
hmm2build HMM corrected.fasta - создание профиля по выравниванию, длина профиля 390hmm2calibrate HMM - калибровка (нормирование весов)
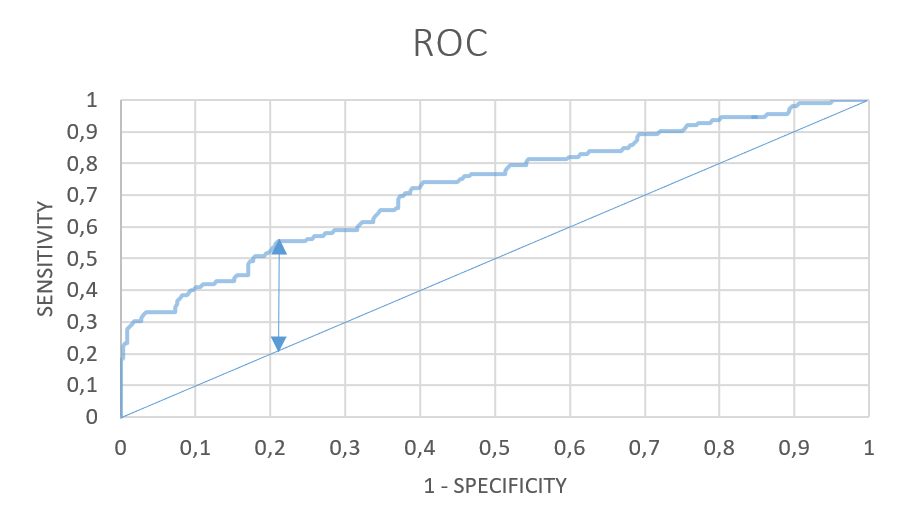
hmm2search -E 0.1 --cpu 1 HMM all.fasta > hmmresult.txt - поиск доменов в последовательностях c E-value находок не более 0.1
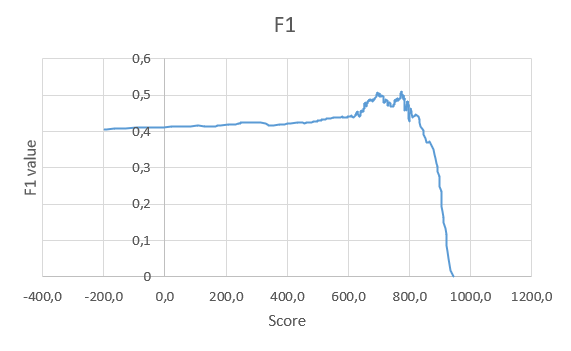
В результате был получен файл, основная информация из которого отражена на листе analyse ранее уже упомянутой таблицы. Столбец true таблицы содержит отметку о том, содержался ли найденный белок в изначальном списке былков с двухдоменной архитектурой. Далее столбцы 1-spec и sens отражают специфичность и чувствительность предсказания пинадлежности белка семейству с двухдоменной архитектурой. Формулы для расчета значений записаны в примечаниях к соответствующим ячейкам и взяты из презентации, предоставленной в указаниях. По этим данным построена ROC-кривая, которая также представлена в таблице. По ROC-кривой предполагалось определить точку с оптимальным соотношением чувствительности и специфичности анализа. Вес находки, которому соответствует эта точка может быть выбран в качестве порогового для установления принадлежности или непринадлежности анализируемого белка семейству с заданной двухдоменной архитектурой. Кроме этого был построен график весов (score) последовательностей с целью определить, после какого значения начинается резкий спад. Это значение также может быть рассмотрено как пороговое. Также в заданиях предлагалось воспользоваться для расчета порогового значения score функцией F1 - средним гармоническим чувствительности и специфичности. Это значение также было рассчитано в соответствующем столбце таблицы, формула, аналогично, сохранена в примечании к ячейке заголовка. Чтобы наглядно показать, как меняется эта функция для разных потенциальных пороговых весов, был построен график F1 от score.
Интерпретация результатов представляет для меня сложность. Во-первых, можно заметить, что программа обнаружила находки с достаточно низкими E-value в 440 из 441 последовательности, хотя, судя по базе данных, в других подсемействах белков домен Stn1 отсутствует (я ожидала 4 дополнительных находки, о которы было упомянуто в начале). График весов не имеет характерной четкой ступеньки и отражает достаточно плавный спад. Значение функции F1 минимально при нулевой чувствительности, то есть при максимально возможном весе находки, далее резко растет и после достижения пика плавно убывает при уменьшении score к минимальным по выборке значениям. Можно отметить, что на графике есть локальный минимум, соответствующий весу находки около 748-749, но если попытаться рассмотреть этот вес как пороговый (границы выделена в таблице синим), нетрудно заметить, что над порогом оказывается очень много находок, не принадлежащих рассматриваемому подсемейству. Итоговый порог я предлагаю провести, согласно анализу ROC-кривой. Чтобы определить оптимальное соотношение параметров, нужно найти точку на кривой, расстояние от которой до диагонали, соединяющей пересечения кривой с осями координат, максимально. Я предполагаю, что это точка с координатами 1-spec -0.78116 sens 0.553571. Ей соответствует вес находки 772.3, что уже ближе к ожидаемому, однако все равно мы наблюдаем много "неправильных" находок выше порога.