Учебный сайт Фоменко Елены
| Главная | Семестры | Проекты | Заметки |
Выбор домена
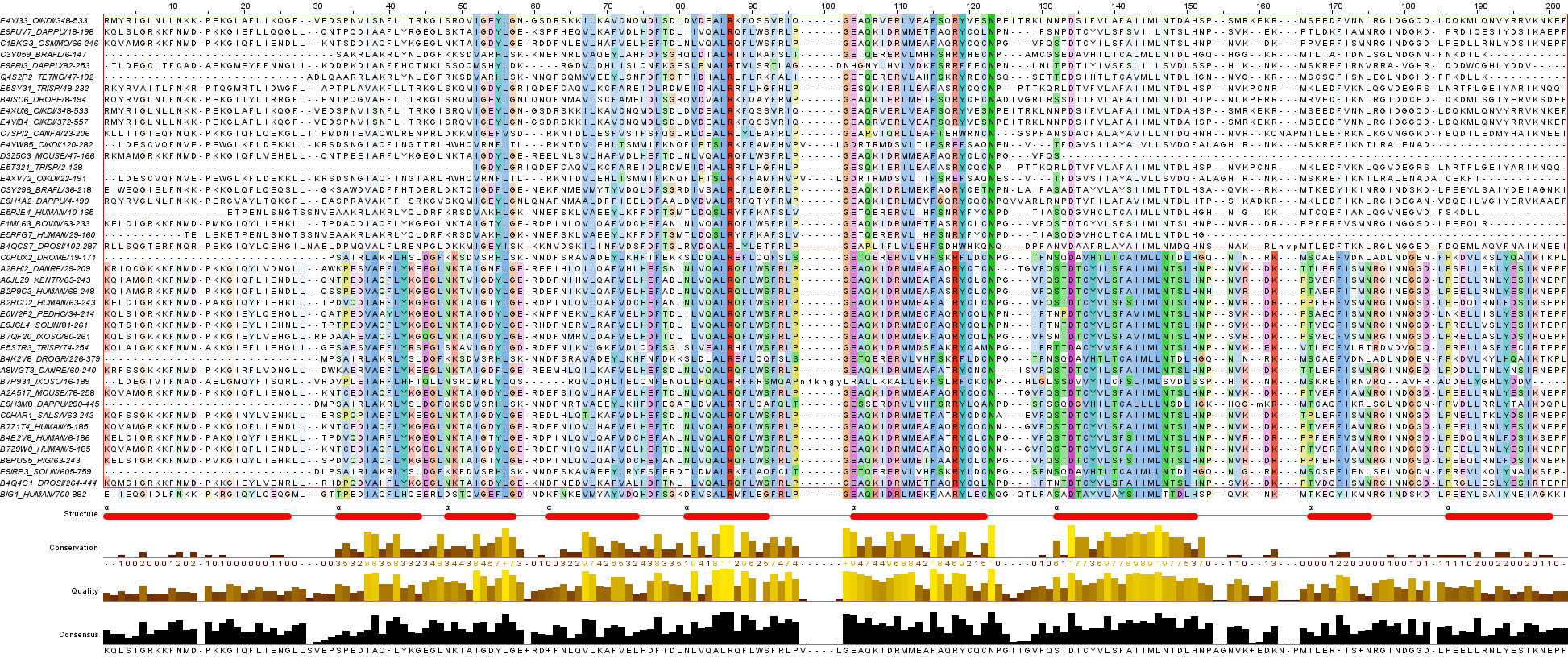
Для работы был выбран домен факторов обмена гуаниловых нуклеотидов (GEFs), Sec7. AC PF01369. Он удовлетворяет всем рекомендуемым ограничениям. GEFs ускоряют обмен ГДФ на ГТФ и таким образом активируют G-белки. C помощью JalView загружено выравнивание из Pfam; раскраска ClustalX и By conservation с порогом консервативности 15%. К последовательности BIG1_HUMAN была добавлена 3D структура (PDB ID 3LTL). Работа сохранена как проект JalView: sec7.jar.
Выбор архитектур
С помощью скрипта swisspfam-to-xls.py и файла,
содержащего информацию об архитектуре всех последовательностей UniProt,
была получена таблица с информацией об архитектуре последовательностей, содержащих домен Sec7,
на основе которой в Excel была составлена сводная таблица.
Затем в таблицу были добавлены колонки с информацией о таксономической принадлежности.
Для этого по идентификаторам отобранных последовательностей в UniProt были получены AC (ID Mapping),
по которым были получены файлы в формате UniProt (Retrieve), которые были использованы для работы скрипта
uniprot-to-taxonomy.py.
Также был добавлен столбец, содержащий информацию о длине домена Sec7.
Sec7 входит в 40 архитектур. Из них выбраны 2:
Sec7 (690 последовательностей)
Sec7, PH (337 послеловательностей),
PH - домен гомологии к плекстрину, характерен для белков, участвующих во внутриклеточном сигнализировании или составляющих
цитоскелет.
Выбор таксона и представителей архитектур
Для изучения указанных выше архитектур был выбран таксон Metazoa (животные), и подтаксоны: клада Ecdysozoa (Линяющие) и тип Chordata (Хордовые). Для однодоменной архитектуры взяла 15 представителей Chordata и только 7 представителей Ecdysozoa, т.к. среди последних было много тех, у которых длина домена слишком мала (в 2 раза меньше нормальной). Для второй архитектуры взяла по 11 представителей с каждого подтаксона. Получилось по 22 представителя для каждой архитектуры. Выборка - на странице selected_seqs в файле Excel.
Чтобы оставить в выравнивании нужные последовательности из двух групп, был использован скрипт filter-alignment.py:
python filter-alignment.py -i sec7.fa -m selected.txt -o filter_alignment_out.fa -a "/"
При этом к выборке был добавлен BIG1_HUMAN, с известной 3D структурой (3LTL), по которой затем была добавлена разметка.
Полученное выравнивание было загружено в JalView и отредактировано
(удалены пустые колонки и несодержательные C- и N- концевые участки).
Затем в нём были выделены группы согласно архитектуре, в каждой из них была выполнена раскраска последовательностей
ClustalX, Conservation (порог 20%). Вот изображение выравнивания.
После редактирования в конечном файле выравнивания JalView содержится 43 последовательности.
{kind=link}
Файлы:
Таблица Excel
Выборка в fasta-формате
Совместное выравнивание всех последовательностей домена
Выравнивание выбранных последовательностей
Построение дерева и выводы
Закодируем доменные архитектуры и выбранные таксоны:
Архитектура: 1 - однодоменная, 2-двухдоменная. Таксоны: E - Ecdysozoa, C - Chordata.
Для реконструкции деревьев использовала два метода - Neighbour-Joining и UPGMA. Первый строит неукорененное дерево без молекулярных часов, а второй - укорененное дерево с молекулярными часами. Для построения была использована программа MEGA. Ниже приведены деревья:
Neighbour-joining
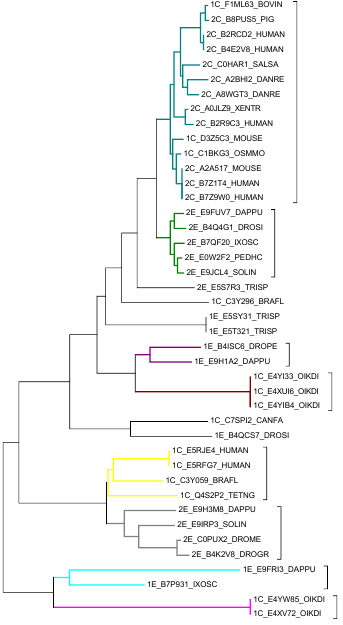
UPGMA

Особых отличий между деревьями я не обнаружила. Ветви, отделяющей архитектуры друг от друга, нет. Несколько ветвей четко разделяют белки по подтаксонам или по архитектурам. Можно предположить, что белок в процессе эволюции изменялся не раз. Можно наблюдать, как у каждого из подтаксонов присходило образование двухдоменного белка, можно наблюдать и разделение белков, произошедшее из-за оразделения подтаксонов, есть случай утраты второго домена. Разными цветами я выделила некоторые заметно отделившиеся группы. Корень, поставленный методом UPGMA, как мне кажется, может и не соответствовать правде.
Файлы: