Учебный сайт Фоменко Елены
| Главная | Семестры | Проекты | Заметки |
1. Аннотацию порфирина в виде SMILES записали в файл 1.smi.
Командой obgen 1.smi > 1.mol строим его структуру и сохраняем формате pdb.
Командой babel -ipdb porphyrin.pdb -omop 1_opt.mop -xk "PM6" создаем входной файл для MORAC.
Запускаем MORAC:
MOPAC2009.exe 1_opt.mop
babel -imopout 1_opt.out -opdb 1_opt.pdb
Сравним исходную структуру и полученную Morac (Рис.1).
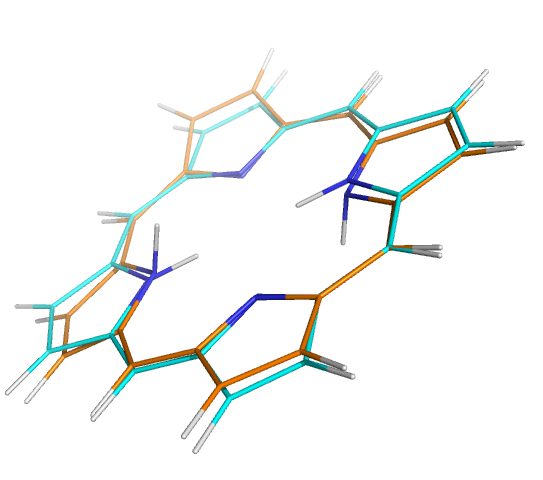
Рис.1 Исходная структура (окрашена оранжевым цветом) и полученная после MORAC (циановая).
Видим, что структура стала плоской (в отличие от немного погнутой исходной).
Входной файл для Mopac: 1_opt.mop
Файл вывода Morac: 1_opt.out
После оптимизации с параметризацией AM1: 2_opt.out/2_opt.pdb
Сравним результаты, полученные при разных параметрах параметризации (Рис.2).
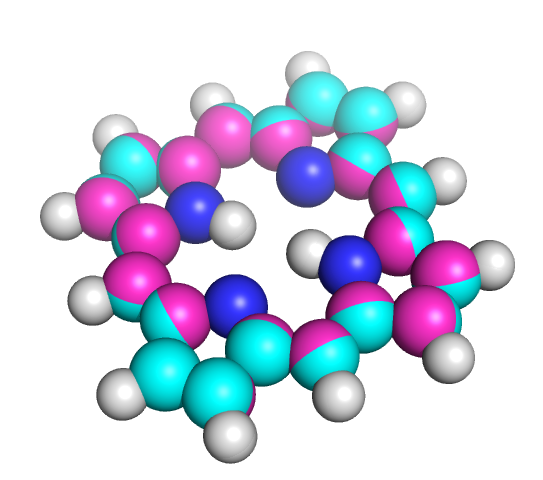
Рис.2 Структуры, полученные разными методами: PM6 (циановая) и AM1 (розовая).
Видим, что при использовании метода AM1 струткура получилась "плотнее", более сжатой (выделена розовым цветом). Метод PM6 считается, однако, более точным, дающим меньше ошибок.
2. Рассчитаем возбужденные состояния порфирина и на основе этих данных прикинем спектр поглощения молекулы.
Для расчёта возбуждённых состояний сделаем копию mop файла из предыдущего задания (1_opt_spectr.mop).
Для указания Mopac о необходимости расчёта возбуждённого состояния добавляем в конец файла:
cis c.i.=4 meci oldgeo
some description
Запускаем Morac:
MOPAC2009.exe 1_opt_spectr.mop
Полученный файл: h1_opt_spectr.out
Находим в конце файла значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.914365 1.914365 1 TRIPLET ????
3 2.266802 2.266802 2 SINGLET ????
4 2.464145 2.464145 2 TRIPLET ????
5 2.825784 2.825784 3 TRIPLET ????
6 3.364386 3.364386 4 TRIPLET ????
7 3.391571 3.391571 3 SINGLET ???? 0.2025 0.2370 0.0011
8 3.669145 3.669145 4 SINGLET ???? 2.3963 2.0275 0.0128
9 3.871984 3.871984 5 SINGLET ???? 1.5412 1.7957 0.0079
На основании этих значений рассчитаем длину волн, при которых происходят эти переходы (count.xls):
ENERGY (EV) wavelength (nm)
1.914365 647,6449933
2.266802 546,9506855
2.464145 503,1477075
2.825784 438,755725
3.364386 368,5156542
3.391571 365,5618319
3.669145 337,9067624
3.871984 320,20507
3. Для молекулы хинона O=C1C=CC(=O)C=C1 определим геометрию как с помощью obgen, так и Мopac (рис.3).
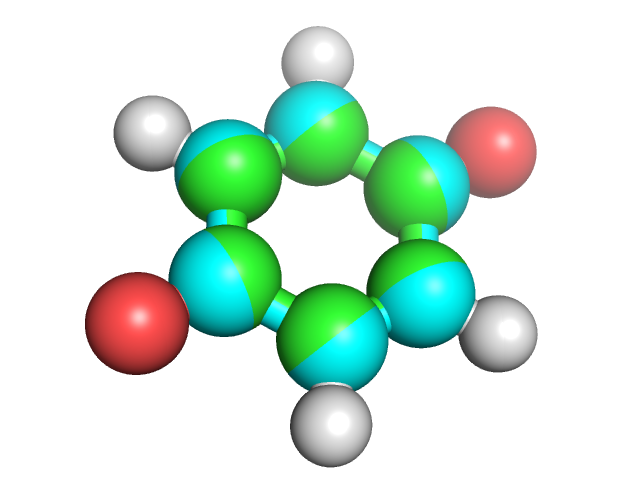
Рис.3 Структура, полученная obgen (зеленая) и Morac (циановая).
Структура, полученная Morac, оказалась немного более "растянутой".
Определим геометрию дианиона этой молекулы. Для начала в первую строчку mop файла добавим слово "CHARGE=-2". Потом укажем, на каких атомах находится отрицательный заряд (два кислорода). Получаем новый mop файл, проделываем с ним то же самое. Сравним результаты Morac c параметризацией PM6 для исходного и модифицированного mop файла (рис.4).
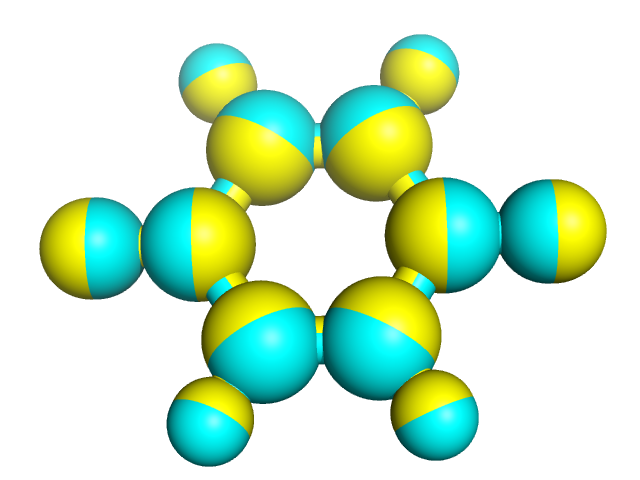
Рис.4 Структура, полученная Morac, исходная (циановая) и из модифицированного mop файла (желтая).
Кажется, что структура дианиона более "сжата" (т.е., уменьшились расстояния между углеродами), но при этом расстояния между кислородами и связанными с ними углеродами увеличились. На самом деле, оказалось, то в исходной структуре это расстояние равно 1,2 Å, в дианионе - 1,3 Å.
4. Известно, что ультрафиолет может превращать тимины в тиминовые димеры,
также известно, что ДНК фотолиаза при облучении ультрафиолетом востанавливает основания тиминов до нормальных.
Нам дан тиминовый димер. Цель - увидеть переход из димера в тимины при возбуждении системы.
Так как вычесления вобуждённых состояний в MOPAC затруденены, мы имитируем возбуждение, ионизируя оба кольца, т.е. указывая заряд системы +2.
И полученое возбуждённое состояние снова оптимизируем при заряде 0.
Результаты оптимизации представлены ниже (рис. 5-7).
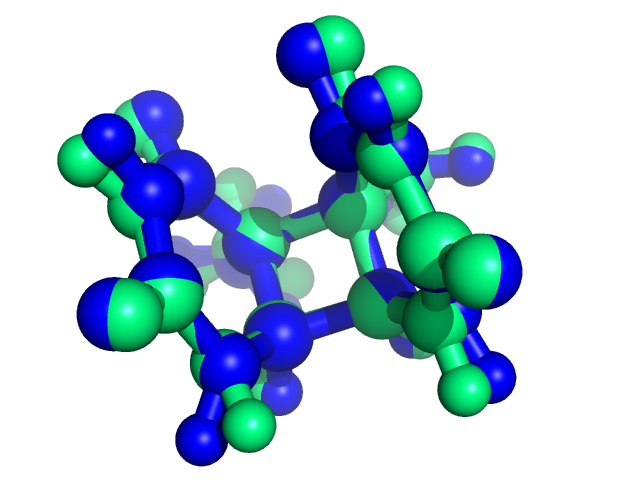
Рис.5 Исходный димер и результат оптимизации его геометрии при заряде системы 0 (окрашены синим и зеленым, соответственно).
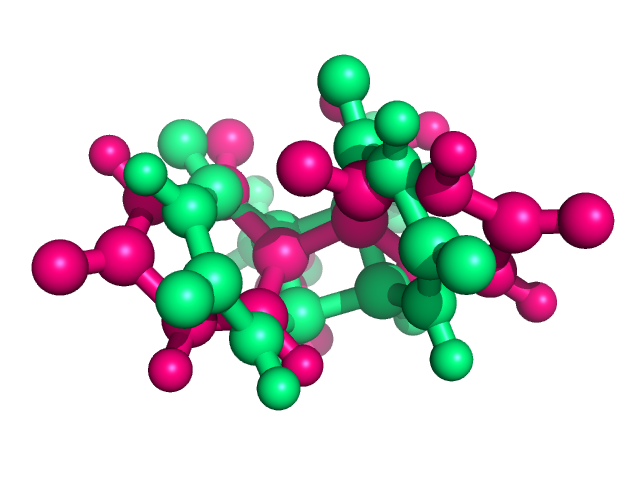
Рис.6 Первая оптимизированная структура и результат ее оптимизации при заряде системы +2 (окрашены зеленым и розовым, соответственно).
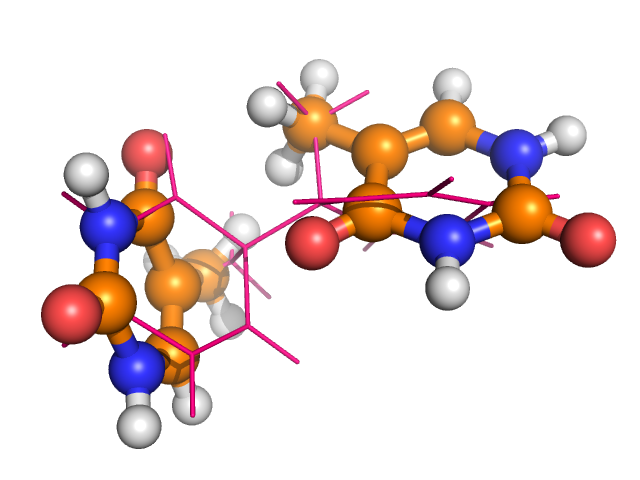
Рис.7 Вторая оптимизированная структура и результат ее оптимизации при заряде системы 0 (окрашены розовым и оранжевым, соответственно).
Сравним энергии этих трех состояний (из out-файлов):
1) -3273.58217 EV
2) -3253.90834 EV
3) -3273.69661 EV
Видим, что при возбуждении энергия поысилась, но при добавлении электронов снова понизилась, при этом не до исходного уровня, а до более низкого. Вероятно, такой переход оказался для системы более энергетически выгодным.
Все файлы лежат в рабочей директории H:\Term8\Practice2