Комплексы ДНК-белок
Первым заданием данного практикума было предсказание вторичной структуры тРНК путем поиска инвертированных повторов и по алгоритму Зукера. Инвертированные повторы мы уже искали с помощью find_pair в прошлом практикуме, теперь нам надо сравнить их с результатами, полученными с помощью команды einverted . В ней требуется задать такие параметры как штраф за гэп, очки за совпадения и несовпадение и другие. Изменяя эти параметры, я получала один и тот же результат:
SEQUENCE: Score 48: 21/32 ( 65%) matches, 3 gaps
1 ggcgcgttaacaaagcggttatg--tag-cggatt 32
|||||||| | || | ||| ||||||
70 ccgcgcaaggcctcagcttggcctgatctgcctaa 36
Далее предсказание вторичной структуры было проведено с помощью алгоритма Зукера, который должен был запускаться с помощью команды mfold в Putty. Но так как эта команда не работала, пришлось воспользоваться сайтом Mobyle @Pasteur. Как и в прошлом методе, здесь тоже можно было вариаровать параметры, но существенное влияние оказал только один - P - отклонение энергии структуры от оптимального. Экспериментально было обнаружено, что лучший результат выходит при Р = 0, он же представлен на картинке ниже.
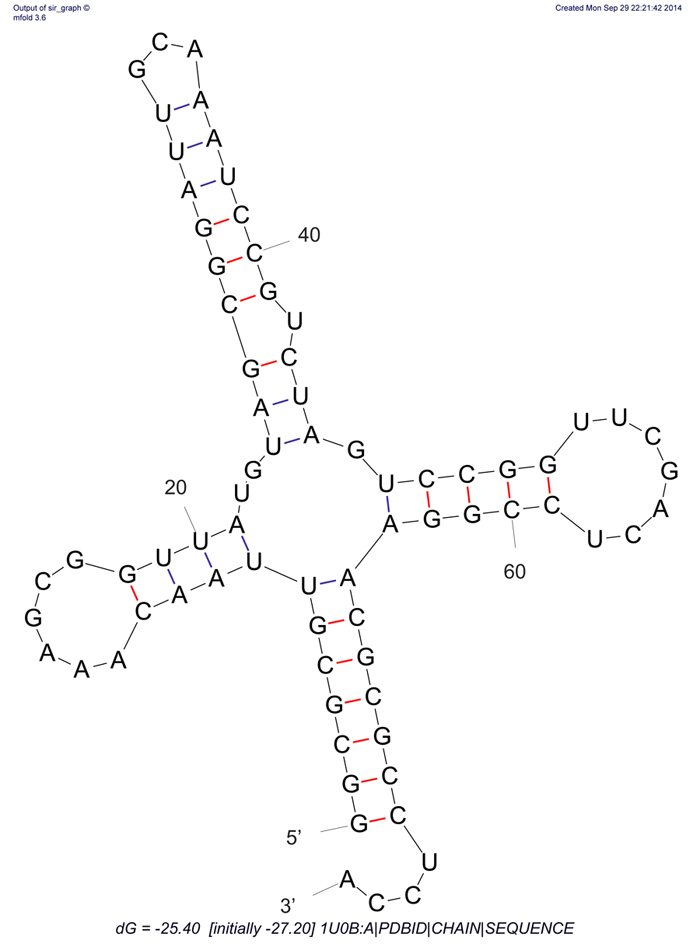
Рисунок 1. Структура тРНК, полученная алгоритмом Зукера.
Таблица 1. Сравнние реальной и предсказаной вторичных структур тРНК.
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5'-1-7-3' 5'-66-72-3' Всего 7 пар |
7 пар из 7 реальных | 7 из 7 реальных |
| D-стебель | 5'-10-12-3' 5'-23-25-3' Всего 3 пары |
Не найдено | 4 из 3 реальных |
| T-стебель | 5'-49-53-3' 5'-61-65-3' Всего 5 пар |
Не найдено | 5 из 5 реальных |
| Антикодоновый стебель | 5'-38-44-3' 5'-26-32-3' Всего 7 пар |
6 пар из 7 реальных | 9 из 7 реальных |
| Общее число канонических пар нуклеотидов | 20 | 13 | 22 |
Из сравнительной таблицы видно, что метод Зукера гораздо эффективнее, чем команда einverted. Причем расхождение в антикодоновом стебле можно объяснить тем, что алгоритм Зукера не умеет распознавать неканонические пары. В оригинальной последовательности имелось 2 неканонические пары нуклеотидов, но алгорит их сместил, образовав петлю и 2 новые канонические пары - как раз то количество, на которое по таблице он превосходит реальную структуру.
Следующим этапом была работа с ДНК с помощью программы Jmol, используя команды define и select within. Нужно сравнить количество контактов разной природы. Ддя этого поделим их на полярные и неполярные. Будем считать полярными атомы кислорода и азота, а неполярными - атомы углерода, фосфора и серы, полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5A. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5A. Результат этой работы занесён в таблицу 2.
Таблица 2. Контакты разного типа в комплексе 1BDT.pdb.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 2 | 29 | 31 |
| остатками фосфорной кислоты | 35 | 41 | 76 |
| остатками азотистых оснований со стороны большой бороздки | 16 | 53 | 69 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 11 | 11 |
Затем нам нужно было получить популярную схему ДНК-белковых контактов с помощью программы nucplot, используя Putty. Результат представлен на рисунке 2 (чтобы увеличить картинку, откройте её в новой вкладке).
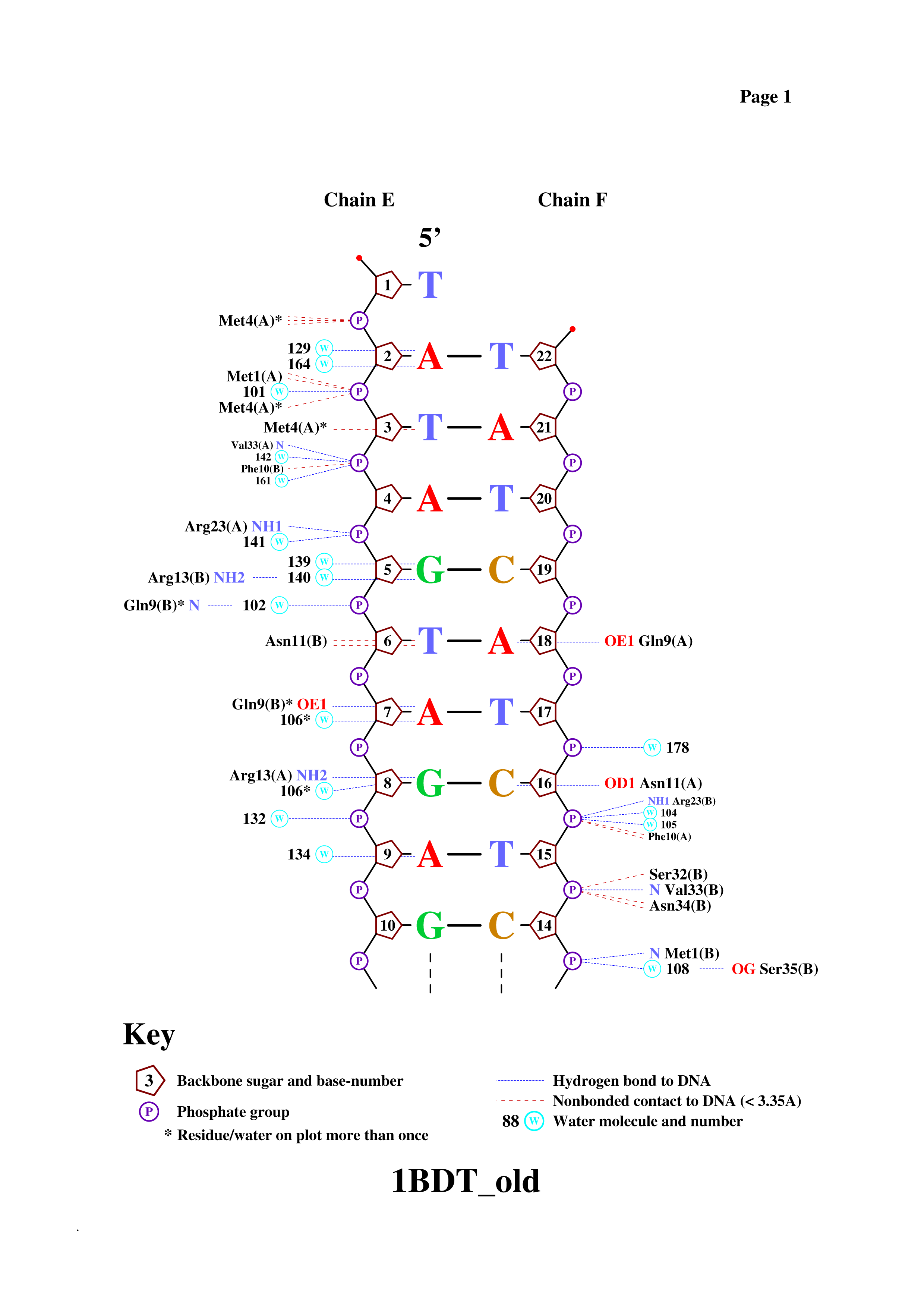
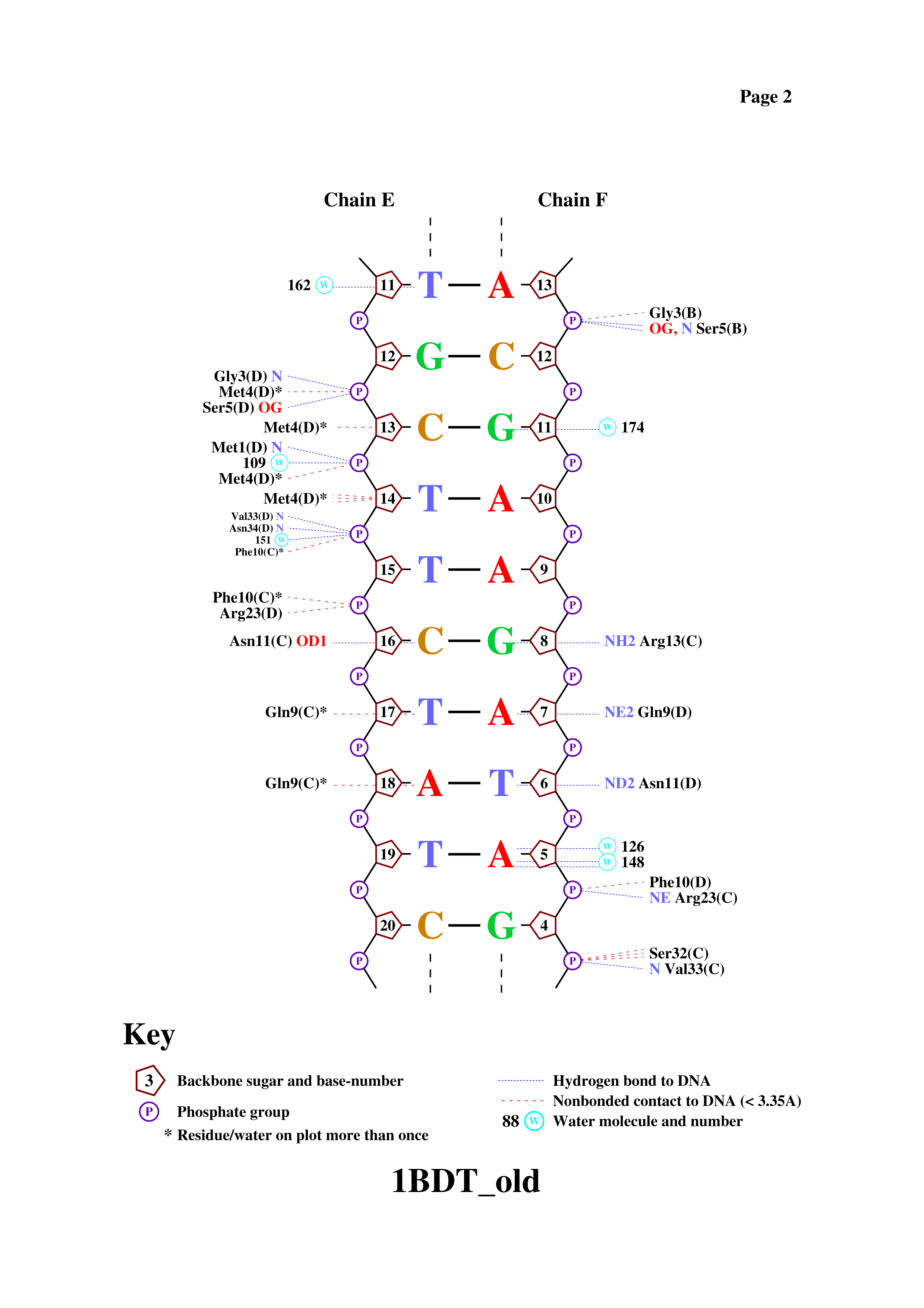
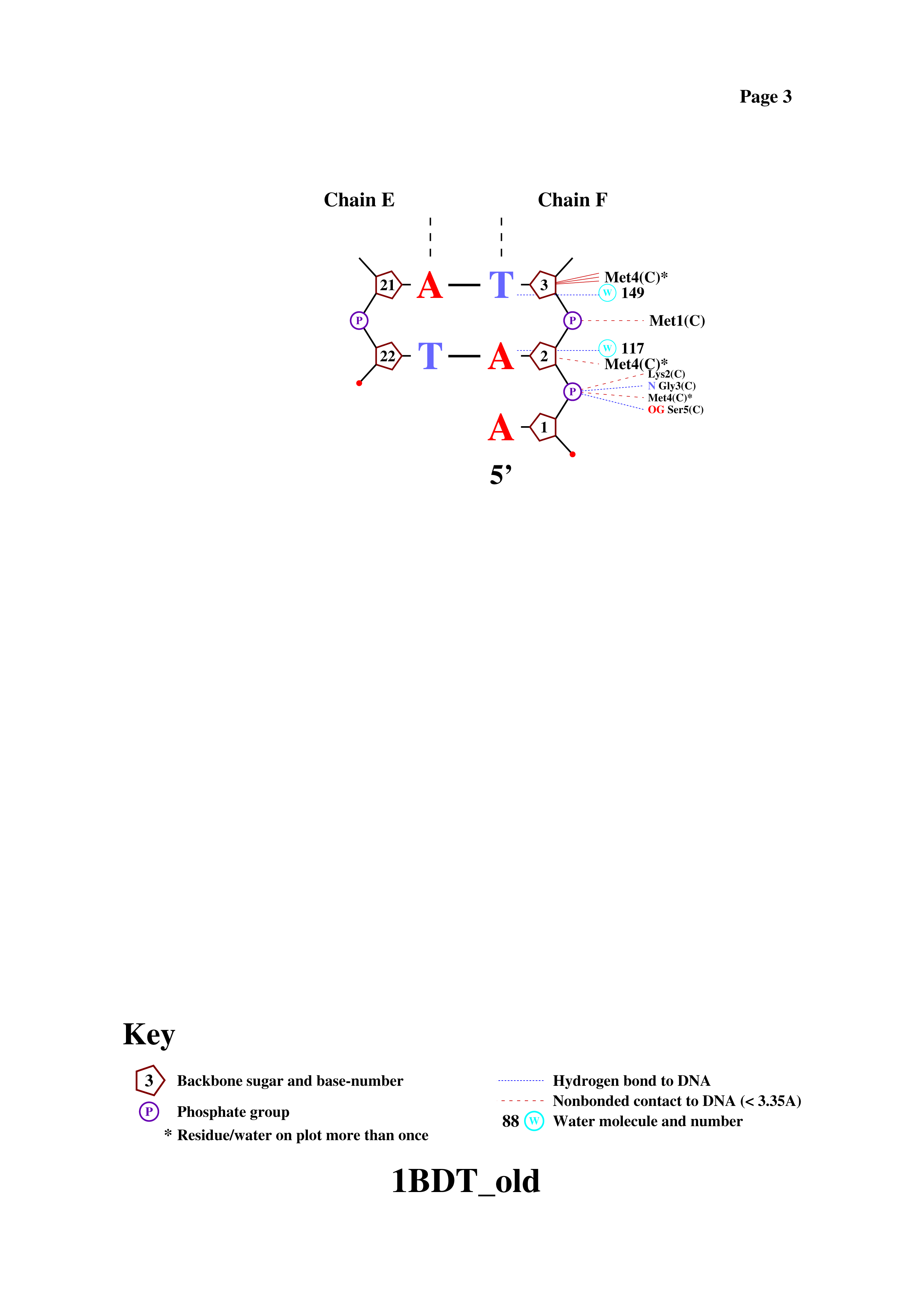
Рисунок 2. Популярная схема ДНК-белковых контактов в структуре 1BDT.
Хоть наибольшее число контактов с ДНК имеет метионин 4, наиболее важным , по моему мнению, для распознавания последовательности ДНК является глутамин 9 цепи С, потому что он соединяет сразу два азотистых основания водородной связью: тимин 17 и аденин 18. Это место в цепи ДНК представлено на рисунке 3.
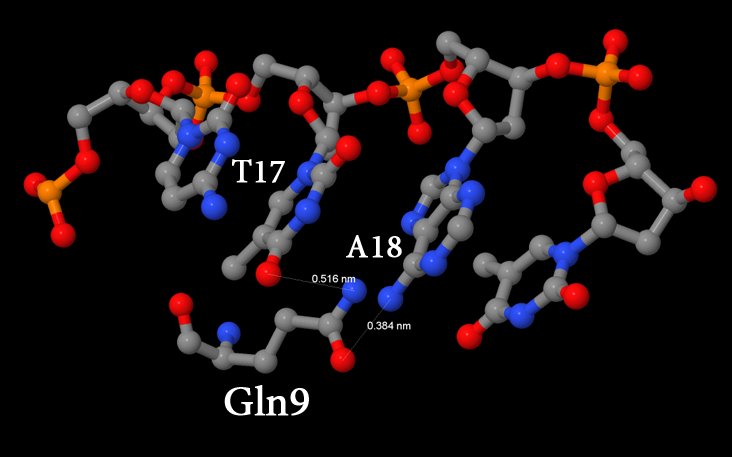
Рисунок 3. Контакты глутамина 9 с ДНК.
Дата последнего изменения: 29.09.2014
