EMBOSS
В данном практикуме мы начали более подробно проходить пакет команд EMBOSS. Для этого давались следующие несколько упражнений, результат выполнения которых можно увидеть ниже.
Программа getorf пакета EMBOSS
Сначала был получен файл с записью D89965 банка EMBL с помощью известной команды entret пакета EMBOSS (entret embl:D89965, полученный файл). Как оказалось, этот файл содержит запись о мРНК из желудка норвежской крысы (Rattus norvegicus), которая содержит ген, кодирующиий рецетор серотонина.
Для дальнейшего выполнения задания нам потребовалось изучение команды getorf. Эта программа находит и выводит последовательности открытых рамок считывания в отдельный файл. Нам нужно было найти набор трансляций всех открытых рамок данной последовательности, которые отвечают следующим требованиям: определены при использовании стандартного генетического кода, имеют длину не менее 30 аминокислотных остатков, начинаются со старт-кодона (то есть с начала последовательности) и заканчиваются стоп-кодоном (до конца последовательности). Учитывая эти факторы, у меня получилась такая команда: getorf -minsize 30 -table 0 -find 1 d89965.entret. Программа нашла 9 рамок считывания, которые можно посмотреть в этом файле, а самой подходходящей записью оказалась пятая:
>D89965_5 [163 - 432] Rattus norvegicus mRNA for RSS, complete cds.
MALMHFQFTFKQFEQRKSIRSTARKARDDFVVVQTADLFHVAFHYGIAQRGLTITSDDHM
AVTAYAYYSCHELTPWLRIQSTNPVQKYGA
Но, хоть эта запись об мРНК из желудка крысы, ссылается она на Uniprot-файл с идентификатором P0A7B8 (/db_xref="UniProtKB/Swiss-Prot:P0A7B8"), а это файл с записью о субъединице HslV АТФ-зависимой протеазы из Escherichia coli (strain K12). Чтобы выяснить, как эти файлы связаны, я скачала Uniprot-файл (entret sw:p0a7b8, полученный файл) и построила выравнивание с файлом, содержащем рамки считывания, при помощи команды needle. В итоге выяснилось, что девятая рамка считывания полностью совпадает с геном E.coli (скопирован фрагмент именно выравнивания, краевые части последовательности, не выравненные, не представлены. Чтобы посмотреть полную картину, скачайте полный файл со всеми выравниваниями.):
HSLV_ECOLI 1 MTTIVSVRRNGHVVIAGDGQATLGNTVMKGNVKKVRRLYNDKVIAGFAGG 50
|||||||||||||||||||||||
D89965_9 1 ---------------------------MKGNVKKVRRLYNDKVIAGFAGG 23
HSLV_ECOLI 51 TADAFTLFELFERKLEMHQGHLVKAAVELAKDWRTDRMLRKLEALLAVAD 100
||||||||||||||||||||||||||||||||||||||||||||||||||
D89965_9 24 TADAFTLFELFERKLEMHQGHLVKAAVELAKDWRTDRMLRKLEALLAVAD 73
HSLV_ECOLI 101 ETASLIITGNGDVVQPENDLIAIGSGGPYAQAAARALLENTELSAREIAE 150
|||||||||||||||||||||||||
D89965_9 74 ETASLIITGNGDVVQPENDLIAIGS------------------------- 98
Этому явлению можно дать вполне логичное объяснение: при отборе клеток желудка для извлечения мРНК могли попасться и бактерии, которые в этом желудке, собственно, и обитают. А далее при секвенировании допустили ошибку из-за случайно попавших E.coli и занесли в базу данных EMBL, а так как в EMBL'е хранится непроверянная информация, то этот огрех сначала никто и не заметил. Но когда заметили, то сделали ссылку на правильную ссылку в Uniprot, потому что там хранится только проверенная информация.
Файлы-списки
В этом задании мы учились применять команды, работающие с файлами-списками. Для начала мы получили файл из банка Swiss-Prot со всеми доступными последовательностям алкогольдегидрогеназ при помощи следующей команды: seqret sw:adh*_*. На выходе получился вот этот fasta-файл.
Следующим шагом было получение файла с универсальными адресами (USA) этих последовательностей: infoseq -only -usa adh.fasta -out usa.txt. Результат выполнения команды можно скачать.
А следующим пунктом уже было получение укороченного списка универсальных адресов - только тех, что были выданы мне, то есть DROGU, DROBU, DROMD, DROHA, MORSE, PAPHA, OCTVU. Для этого я сделала txt-файл с этими адресами, с которым и проводилась дальше работа. Чтобы выбрать определённые записи, нужно было воспользоваться командой grep. Вот такая команда получилась у меня: grep -f organisms.txt usa.txt >> narrow_usa.txt. Результатом является вот этот txt-файл.
И последним этапом было получение fasta-файла со последовательностями из моего списка. Для этого была использована следущая команда: seqret @usa_narrow.txt sequences.fasta. Итоговый документ с результатом можно посмотреть по этой ссылке.
Случайная модель для оценки достоверности выравнивания
Для оценки достоверности вывода о реальности гомологии последовательностей на основе веса их выравнивания было проведено сравнение со случайной моделью.
Сначала я выбрала две алкогольдегидрогеназы из моего списка (см. предыдущее задание): Drosophila hawaiiensis (fasta-файл) и Papio hamadryas (fasta-файл). Далее с помощью команды shuffleseq было сделано 100 случайных перемешиваний аминокислотной последовательности белка Drosophila hawaiiensis: shuffleseq ADH_DROHA.fasta. Полученный файл с перемешанными последовательностями можно посмотреть тут.
Далее были построены выравнивания неперемешанной последовательности ADH1B_PAPHA со всеми перемешанными вариациями ADH_DROHA и с оригинальной с помощью команды water. Полученные файлы можно посмотреть: выравнивание с оригиналом и выравнивания с "перемешанными" последовательностями. И затем, с помощью команды grep (grep '# Score:' xxx.water | sed 's/# Score: //g' > xxx_xxx.txt) были получены списки весов этих выравниваний, которые можно скачать: вес оригинального и веса перемешанных выравнивания. На основе этих данных была построена гистограмма распределения полученных весов (рисунок 1) с шагом в 3 единицы, который, по-моему мнению, наиболее ярко отражает картину.
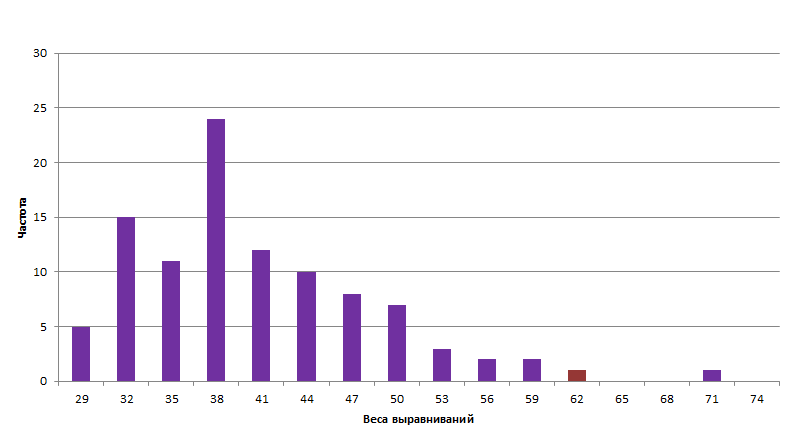
Рисунок 1. Гистограмма распределения весов выравниваний последовательности алкогольдегидрогеназы Papio hamadryas и с сотней "перемешанных" последовательностей алкогольдегидрогеназы Drosophila hawaiiensis. Бордовым выделен столбик, в который входит вес выравнивания с оригинальной последовательностью. Рисунок получен с помощью Microsoft Excel и Paint.
Из гистограммы видно, что вес "реального" выравнивания находится между значениями 62 и 65, а точнее он 62,5. Это единственное значение в этом диапозоне и, к тому же, сильно отличается от большинства других значений "весов" выравниваний, так что можно считать, что настоящее выравнивание достоверно.
Далее, с нуклеотидными последовательностями этих генов были проведены те же операций: shuffleseq (опять же для гена алкогольдегидрогеназы Drosophila hawaiiensis, water и grep. В результате была также получена гистограмма распределения весов с шагом в 10 единиц, которая представлена на рисунке 2.
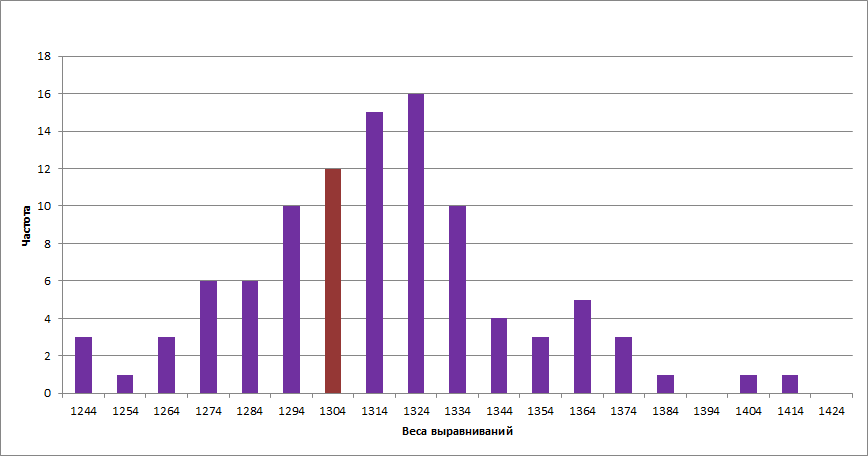
Рисунок 1. Гистограмма распределения весов выравниваний нуклеотидной последовательности алкогольдегидрогеназы Papio hamadryas и с сотней "перемешанных" последовательностей алкогольдегидрогеназы Drosophila hawaiiensis. Бордовым выделен столбик, в который входит вес выравнивания с оригинальной последовательностью. Рисунок получен с помощью Microsoft Excel и Paint.
Из гистограммы видно, что вес реального выравнивания располагается между значениями 1304 и 1314, а точнее он равен 1313,5. В этом случае он отличается от перемешанных выравниваний меньше, можно даже сказать, что он приближен по значениям к большинству значений непарных выравниваний, что позволяет усомниться в его достоверности. Из этого можно сделать вывод, что при перемешивании нуклеотидных последовательностей шанс получения подходящего по весу выравнивания больше, чем при перемешивании аминокислотных последовательностей.
Дата последнего изменения: 17.10.2014
