Standalone BLAST
Поиск некодирующих РНК (misc_RNA), аннотированных в одном штамме, в геноме другого штамма
Для выполнения этого задания сначала мы получили файл, содержащий последовательности со всеми misc_RNA штамма Bacillus_subtilis_168_uid57675. Для этого из frn-файла, содержащего все аннотированные РНК этого штамма, был получен лист-файл при помощи команды infoseq. При помощи команды Grep был получен файл, содержащий только адреса последовательностей misc_RNA: miscRNA.txt. И уже с помощью комнады seqret был получен fasta-файл, содержащий сами последовательности РНК.
Искать некодирующие последовательности, закодированные в первом штамме, мы будем искать в Bacillus_subtilis_spizizenii_TU_B_10_uid73967. Были скачены два файла с его геномом: gbk-файл и fasta-файл. Используя локальный BLAST и геном второго штамма как базу данных, мы искали гомологов данных РНК. С помощью комнады blastn (blastn -task megablast -query miscRNA.fasta -db NC_016047.fna -out task1.out -outfmt 7 -num_alignments 1) был получен файл с информацией о всех выравниваниях. На основе этого файла была составлена таблица Excel, где для каждой misc_RNA указаны её номер, цепь (прямая или обратная относительно записи RefSeq) и координаты начала и конца лучшего найденного гомолога.
Поиск гомологов РНК Bacillus subtilis в геноме другой бактерии
В этом задании в качестве безы данных использовался геном бактерии Bacillus cereus AH187 uid58753, в качестве исходных данных тот же самый штамм. Поиск проводился с помощью трёх различных программ и были получены три выходных файла: стандартный megablast, стандартный blastn и blastn со специфическими параметрами (-word_size 4, -penalty -1, -reward 1). На основе этих результатов была составлена таблица Excel, в которой для каждого случая поиска было указано количество найденных гомологов с e-value < 0.001.
Поиск неправильно аннотированных генов программой blastx
В третьем задании практикума был проведён поиск гомологов misc_RNA из предыдущих заданий в скачанных белковых последовательностях с помощью программы blastx. В качестве базы данных использовался файл с последовательностями предсказанных белков штамма Bacillus subtilis spizizenii TU B 10 uid73967. Blastx на вход принимает файл с нуклеотидными последовательностями и ищет среди белковых. Для осуществления поиска был определён e-value не больше 0,001 и составлена команды: blastx -query miscRNA.fasta -db NC_016047.faa -out blastx.out -evalue 0.001 -outfmt 7 -num_alignments 1. Выходной файл - task3.out. В итоге гомологи были найдены для РНК под номерами 1, 25, 41, 46, 53, 57, 58, 61, 62; но только 1-ый и 46-ой белки были реальными, а не гипотетическими (серил-тРНК синтаза - 1 и аспартокиназа - 46).
Зависимость времени работы blastn от длины затравки
Для того, чтобы проанализировать зависимость времени работы blastn от длины затравки, была проведена серия поисков гомологов РНК штамма Bacillus_subtilis_168_uid57675 в геноме Bacillus cereus AH187 uid58753 в качестве базы данных. Длина слова варьировалась от значения 4 до 16. Команда, которая использовалась для определения времени: time blastn -task blastn -query miscRNA.fasta -db NC_011658.fna -out test.blastn -word_size x, где х = [4;16]. Результаты анализа показаны в таблице 1 и на рисунке 1.
Таблица 1. Зависимость времени (в секундах) от длины затравки.
| Длина затравки | Время в сек |
| 4 | 79.526 |
| 5 | 20.799 |
| 6 | 6.011 |
| 7 | 2.226 |
| 8 | 1.282 |
| 9 | 1.103 |
| 10 | 0.899 |
| 11 | 0.815 |
| 12 | 0.796 |
| 13 | 0.783 |
| 14 | 0.747 |
| 15 | 0.713 |
| 16 | 0.677 |
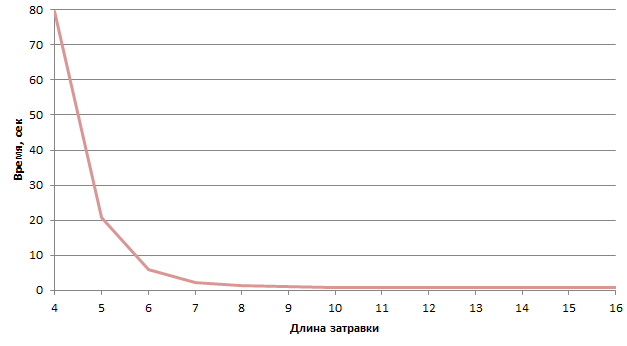
Рисунок 1. Зависимость времени (в секундах) от длины затравки.
Заметно, что при увеличении длины затравки уменьшается время поиска. Возможно, это связано с тем, что количество гомологов, которое находится меньше (так как ужесточаются рамки поиска) и, следовательно, меньше времени требуется на обрабатывание данных и их записывание в файл.
Дата последнего изменения: 12.10.2014
