Программе MODELLER для моделирования структуры белков, в качестве входных данных нужны: управляющий скрипт, файл pdb со структурой-образцом, файл выравнивания с дополнительной информацией.
Выравнивание
С помощью программы ClastalW построено выравнивание последовательности из структуры ID: 1lmp и предложенного белка LYS_BPPHV (UniProt), в качестве формата выходного файла был сразу указан PIR:смотреть.
Модификация файла выравнивания
Переименовала последовательность в файле выравнивания:| Было | Стало |
| >P1;uniprot|P37712|LYSC_CAMDR | >P1;seq |
| >P1;1LMP|PDBID|CHAIN|SEQUENCE | >P1;1lmp |
После имени последовательности моделируемого белка надо добавить строчку:
sequence:ХХХХХ::::::: 0.00: 0.00эта строчка описывает входные параметры последовательности для modeller. После имени последовательности белка-образца добавить:
structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00эта строчка описывает, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот в структуре, идентификатор цепи и т.д. В конце каждой последовательности надо добавить символы:
/.Символ "/" означает конец цепи белка. Точка указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки).
файл
Модификация файла со структурой
Надо удалить всю воду из структуры (в текстовом редакторе), всем атомам лиганда присвоить один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицировать имена атомов каждого остатка, добавив в конец буквы A, B, C. Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д. После модификации имен атомов изменить номера остатков на 130.Пример:
| Было | Стало |
| HETATM 1014 O7 NAG 130 | HETATM 1014 O7A NAG 130 |
| HETATM 1015 C1 NAG 131 | HETATM 1015 C1B NAG 130 |
Измененный файл сохранен как 1lmp_now.ent.
Создание управляющего скрипта lys_bpphv.py
lys_bpphv.pyВ скрипте указано:
что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4);
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема);
атомы белка, образующие водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов (длина данного белка 226 а.о., а белка-модели - 129, поэтому номера а.о. лиганда несколько изменились);
параметры взаимного расположения атомов пары описаны в строчке 9-10. 3 точки могут однозначно расположить сложную структуру в пространстве, поэтому мы выбираем водородные связи как источник данных точек;
что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами, строчка 12;
что имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании);
что число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15
что пора строить модель, строчка 16
Запустим исполнение скрипта командой
mod9v7 lys_bpphv.py &
Поскольку в выравнивании есть очень большая вставка, то получившийся результат не очень хороший, т.к. существование такой большой петли в растворе маловероятно:
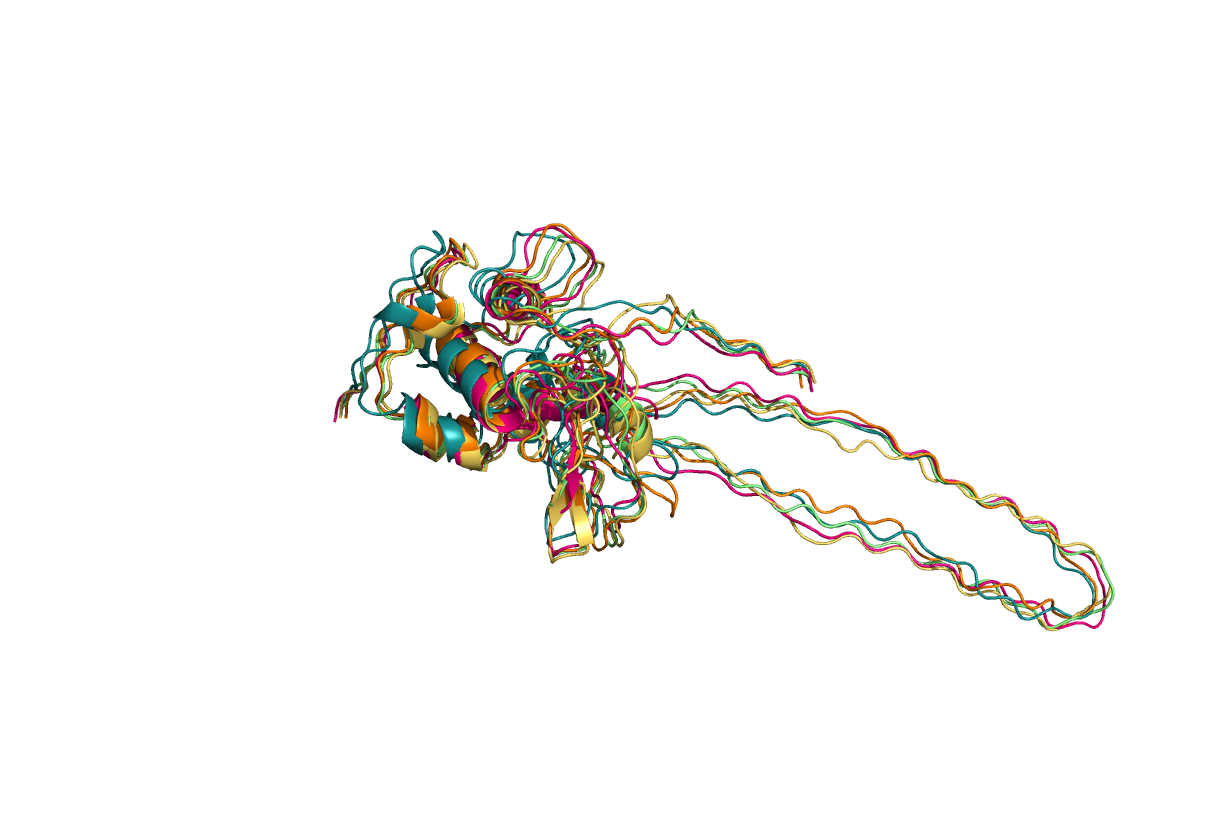
Но эта петля находится далеко от активного центра, поэтому, в принципе, нам не очень важно знать ее точную 3D структуру.
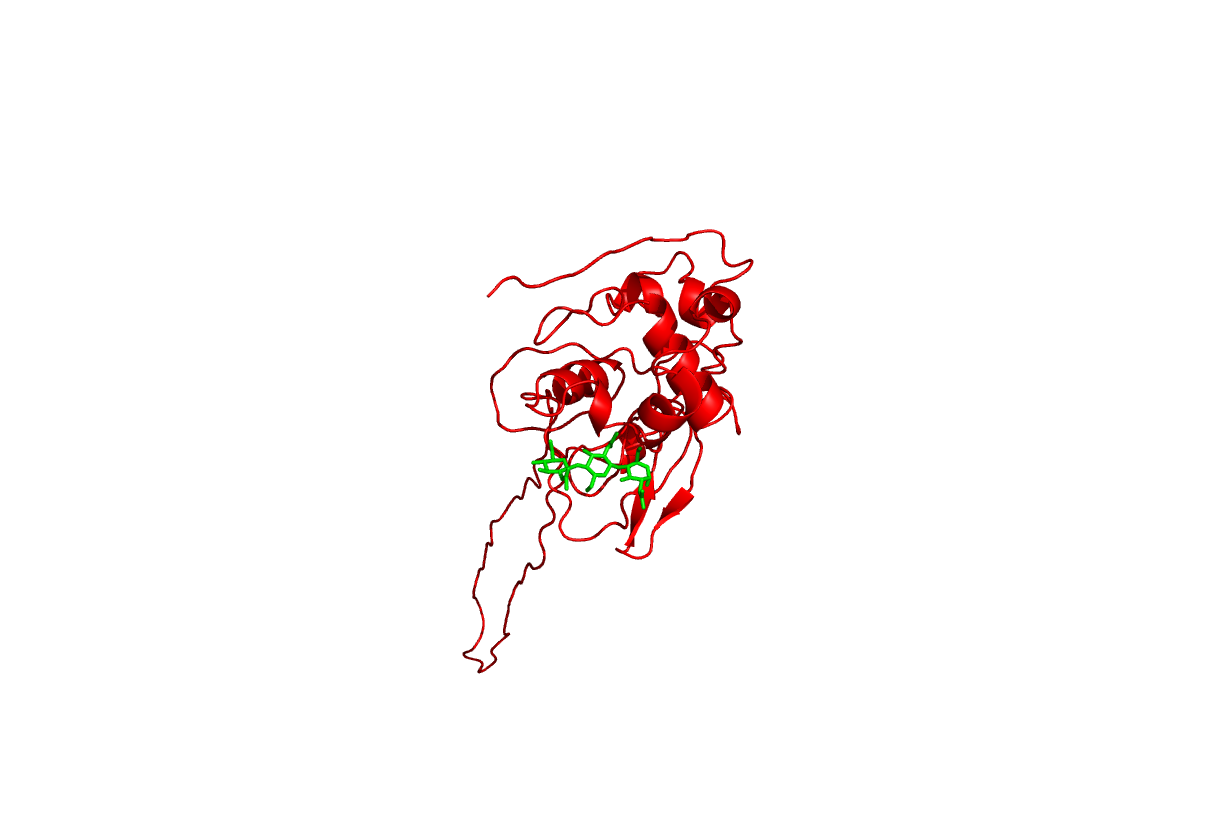
Проверка правильности модели с помощью WHATIF:
-Secondary Structure, symmetry and accessibility
Вторичная струтура несколько отличается от 1LMP, поскольку выравнивание не очень хорошее.
-Interatomic bumps
По умолчанию WHATIF считает, что расстояние между атомами, не свзанных водородными связями, должно быть не короче, чем 2*Ван-дер-Ваальсовый радиус - 0.4Å; для связанных - не короче 0.55Å. Если атомы располагаются ближе, то это указывается в выдаче. Полученная струтура имеет очень много наложений (511) по сравнению с 1LMP (всего 24).
-Fine Packing Quality Control
Упаковка остатков по качеству такая же, как и у 1LMP.