Ресеквенирование. Поиск полиморфизмов у человека.
0. Полученные файлы.
> Чтения: chr11.fastq
> Хромосома человеческого генома: chr11.fasta
Их можно найти в директории /nfs/srv/databases/ngs/mariagur
1. Анализ качества чтений.
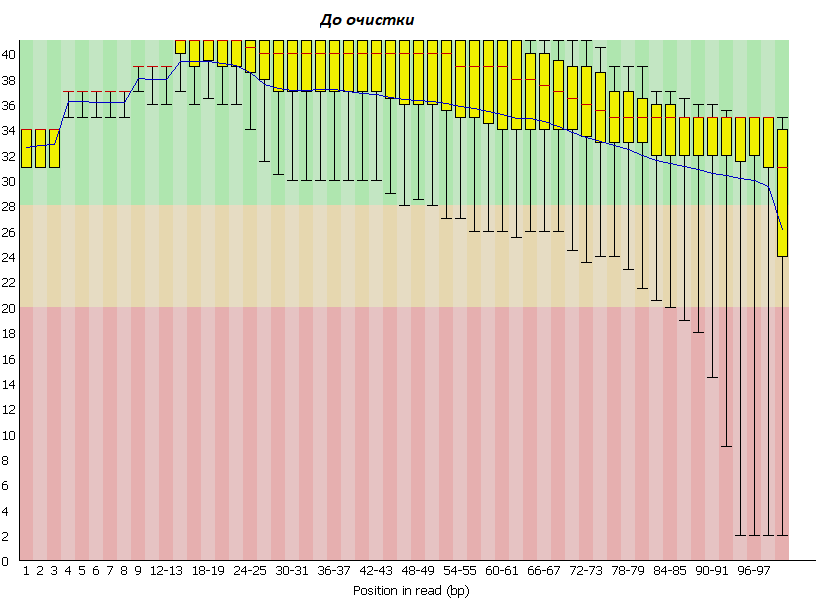
При помощи программы FastQC был проведен контроль качества ридов.
Её результат был выдан в качестве страницы html. Из него видно, что качество
прочтений достаточно хорошее, так как синяя линияя(показывающяя среднее значение) лежит в интервале от 28 до 40 и лишь к концу попадает
в оранжевый интервал.
2. Очистка чтений.
С помощью программы Trimmomatic была произведена очистка чтений. Удалялись прочтения из конца с качеством ниже 20,а также прочтения длиной менее 50.
В руководстве были найдены команды для такой чистки:
TRAILING:
MINLEN:
В итоге запущенная команда выглядела так: java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr11.fastq chr11_trim2.fastq TRAILING:20
MINLEN:50
Очищенные прочтения также были проанализированы с помощью FastQC.Полученный результат.

Количество чтений до чистки: 4198
Количество чтений после чистки: 4064
Из картинок, выданных FastQC, видно, с конца убралось достаточное количество ридов, тем самым
значительно улучшив показатель качества. Мне каежтся, что это связано с тем, что в конце уже накопилось довольно много ошибок, которые понижали качество прочтений.
3. Картирование чтений.
Файл выданной программой Hisat2
Вызванные команды:
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
hisat2-build chr11.fasta chr11
hisat2 -x chr11 -U chr11_trim.fastq -S ex3.sam --no-softclip --no-spliced-alignment
4. Анализ выравнивания
| Вызванные команды: | Что делают: |
| samtools view -b ex3.sam -o ex4.bam | Переведение выравненных с референсом чтений(файл ex3.sam) в бинарный формат .bam |
| samtools sort ex4.bam -T ex4.txt -o ex4_sort.bam | Сортировка выравненных с референсом чтений(в .bam формате) по координате в референсе начала чтения |
| samtools index ex4_sort.bam | Индексация отсортированного .bam файл |
| samtools idxstats ex4_sort.bam > out.txt | Показывает сколько чтений откартировано на геном |
Из 4063 откартировано на геном 4048. 15 прочтений не откартировались.
5. Поиск SNP и инделей.
| Вызванные команды: | Что делают: |
| samtools mpileup -uf chr11.fasta ex4_sort.bam -o ex5_snp.bcf | Создание файла с полиморфизмами в формате .bcf |
| bcftools call -cv ex5_snp.bcf -o ex5_snp.vcf | Создание файла со списком отличий между референсом и чтениями в формате .vcf |
Было найдено 3 полиморфизма из файла .vcf:
| Номер | Координата | Тип полиморфизма | Референсная последовательность | Чтения | Глубина покрытия | Качество чтений |
| 1 | 17408305 | Замена | G | C | 14 | 103.008 |
| 2 | 116628401 | Замена | T | C | 92 | 187.009 |
| 3 | 116657590 | Вставка | CAAA | CAAAA | 62 | 147.467 |
Всего было получено 21 snp и 2 инделя.
6. Аннотация SNP.
/nfs/srv/databases/annovar/annotate_variation.pl -out refg -build hg19 ex6.avinput /nfs/srv/databases/annovar/humandb/
Было получено, что 3 SNP относятся к UTR3, 5 лежат в экзонных областях, а 12 в интронных. Эти SNP лежали в границах 3х генов: KCNJ11, BUD13, ZPR1.
/nfs/srv/databases/annovar/annotate_variation.pl -filter -out out_rs -build hg19 -dbtype snp138 ex6.avinput /nfs/srv/databases/annovar/humandb/
Из 21 SNP - 19 имеют rs.
/nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out out_1000g ex6.avinput /nfs/srv/databases/annovar/humandb/
Среднее значение частоты найденных snp составляет 0,35; минимальное - 0,0034; максимальное - 0,88;
/nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -out out_clin ex6.avinput /nfs/srv/databases/annovar/humandb/
Клиническая аннотация выдала 2 snp. Первый - непатогенный и не специфичный, второй - связан с сахарным диабетом 2ого типа/неонатальным диабетом.
/nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out ex1 -dbtype gwasCatalog -out out_gwas ex6.avinput /nfs/srv/databases/annovar/humandb/
Было получено 6 snp с клиническим значением. Два из них относились к сахарному диабету 2ого типа,один - метаболический синдром.(подробнее см. сводную таблицу)
Сводная таблица со всеми snp: excel таблица
Вывод: Из таблицы видно, что две мутации в экзонных областях(координаты в первом столбце выделены красным), вероятно связаны с сахарным диабетом. Это подтверждается аннотациями из таких баз данных, как Clinvar и Gwas. Также одна замена в экзонной области(координата выделена зеленым) привела к метаболическому синдрому, который также непосредственно связан с диабетом 2-ого типа. Однако для него не было найдено аннотации в Clinvar.