Подготовка чтений состояла из двух стадий: анализа качества чтений и очистки чтений.
Анализ качества чтений выполнялся при помощи программы FastQC [1]. FastQC выполняет ряд тестов, позволяющих составить "первое впечатление" о качестве исследуемых данных. На выходе программа выдает html-файл с описанием.
>Команда: fastqc chr11.fastq
>Результат доступен по ссылке: FastQC-report
>Результат доступен по ссылке: FastQC-report
Очистка чтений была сделана с помощью программы Trimmomatic [2], которая разработана для обработки данных парноконцевого секвенирования Ilumina. Были вырезаны нуклеотиды с качеством ниже 20, а также все чтения длиной меньше 50 нуклеотидов.
TRAILING: Удаляет участки в конце с качеством ниже заданного
MINLEN: Удаляет риды длинной меньше заданной
>Команда: java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr11.fastq trimmed.fastq LEADING:20 TRAILING:20 MINLEN:50
Таким образом было вырезано 134 рида из 4198, что скорее всего указывает на высокое качество изначальных данных. Для проверки этого предположения полученный файл trimmed.fastq был проанализирован с помощью программы FastQC.
>Команда: fastqc trimmed_fastqc
>Результат доступен по ссылке: FastQC-report
На изображенниях ниже (рис.1, рис.2) представны результаты работы программы в графическом интерфейсе.
>Результат доступен по ссылке: FastQC-report
Сравнение результатов до и после очистки некачественных ридов:
1. Параметр "per base sequence quality"(рис.1). Так как были удалены все риды длиной меньше 50, на "очищенной" схеме все риды располагаются в зеленой области. Катинка распределения остальных тоже немного меняется, что указывает на то, что были удалены ненадежные прочтения.
2. Параметр "Sequence Duplication Levels" - как видно из рисунка 2, не все параметры ощутимо меняются при очистке чтений. Например, в данном случае, уровень дупликаций почти не меняется.
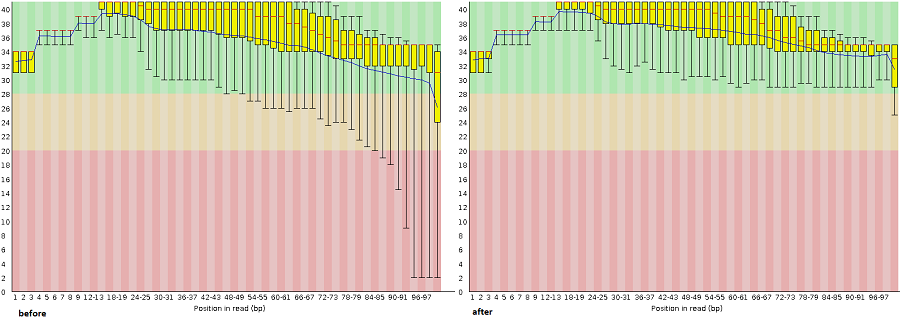
Рис.1 Сравнение параметров "per base sequence quality" до и после обработки ридов в программе Trimmomatic.
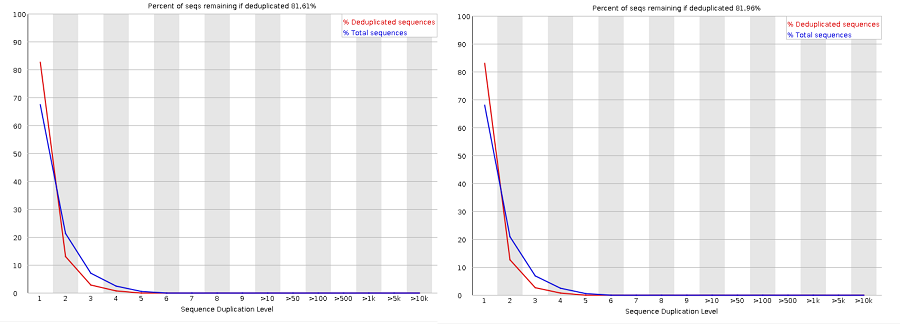
Рис.2 Сравнение параметров "Sequence Duplication Levels" до и после обработки ридов в программе Trimmomatic.
Часть 2. Картирование чтений
Картирование чтений производилось с помощью программы Hisat2 [3] и включало в себя индексацию референсной последоваельности и построение выравниваний прочтений и референса в формате .sam.
>Пусть к программам, необходимым для запуска Hisat2: export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
>Индексирование референса: hisat2-build chr11.fasta chr11
>Построение выравниваний: hisat2 -x chr11 -U trimmed.fastq -S align.sam --no-softclip --no-spliced-alignment
>Индексирование референса: hisat2-build chr11.fasta chr11
>Построение выравниваний: hisat2 -x chr11 -U trimmed.fastq -S align.sam --no-softclip --no-spliced-alignment
Анализ выравнивания
Были последовательно выполнены следующие команды:
| Команда | Функция |
| samtools view -b align.sam -o align.bam | Переводит выравнивание (референс + чтения) из формата .sam в бинарный формат .bam |
| samtools sort align.bam -T align.txt -o align_sorted.bam | Сортирует выравнивание по коортинате референса в начале чтения |
| samtools index align_sorted.bam | Индексирует полученный после сортировки .bam-файл |
| samtools idxstats align_sorted.bam >idxstats.txt | Подсчитывет количество чтений, откартированных на геном и выводит результат в файл idxstats.txt |
В результате проведенных опрераций 4049 чтений картировались на хромосому, 15 - нет. Для анализа покрытия я воспользовалась командой depth. На выходе был получен файл с покрытиями нуклеотидов по позициям. Проанализировав данные в Excel я выяснила, что покрытие на нуклеотид принимает значения от 1 до 165, среднее составляет 17, а медиана - 23,8. На рис. 3 приведена гистограмма распределения покрытий по нуклеотидам (значения предварительно были отсортированы для наглядности).
С помощью Genom Browser[4] я определила границы экзона для выбранного мной нуклотида с высоким покрытием (116628512, покрытие 185): 116628482-116628666.
>Анализ покрытий всех нуклеотидов: samtools depth align_sorted.bam > depth.txt
>Анализ покрытий с учётом границ экзона: samtools depth align_sorted.bam -r chr11:116628482-116628666 > depth-exon.txt
>Анализ покрытий с учётом границ экзона: samtools depth align_sorted.bam -r chr11:116628482-116628666 > depth-exon.txt
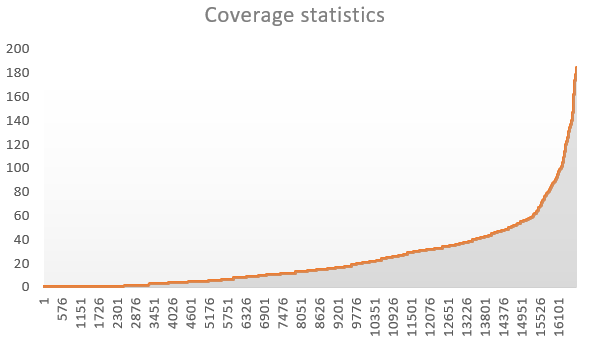
Рис.3 Распределение покрытий по нуклеотидам
При анализе покрытия с указанием определенного экзона среднее значение покрытия получилось 143,2 (медиана 150). График выглядит немного странно, но пусть будет:
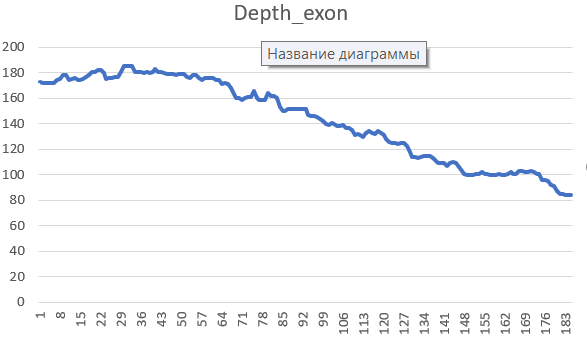
Рис.4 Распределение покрытия в экзоне с хорошим покрытием
Часть III: Анализ SNP
Анализ однонуклеотидных полиморфизмов (SNP = Single Nucleotide Polymorphism) включает в себя поиск SNP и инделей, а затем аннотацию SNP.
Поиск SNP и инделей
| Команда | Функция |
| samtools mpileup -uf chr11.fasta align_sorted.bam -o snp.bcf | В файл snp.bcf занисываются найденные полиморфизмы |
| bcftools call -cv snp.bcf -o snp.vcf | Создаётся файл vcf, который представляет собой таблицу полиморфизмов относительно референса |
В полученном vcf-файле содержится 23 полиморфизма, из которых 21 - замены, 2 - индели. Подробное описание 3х из них примедено в таблицк ниже:
| Координата | Тип | Референс -> чтения | Глубина покрытия | Качество чтений |
| 116649538 | Замена | А -> G | 11 | 86.0076 |
| 116653145 | Замена | C -> T | 2 | 4.76921 |
| 116650454 | Делеция | ATCTCT -> ATCT | 22 | 196.468 |
Аннотация SNP
Аннотация полиморфизмов (только snp) по разным базам данных производилась с помощью команды annovar [5]. Последовательность действий представлена в таблице ниже:
| Команда | Функция | Тип аннотации |
| - | Из snp.vcf вырезаны индели | |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp.vcf > snp.avinput | Перевод таблицы snp в необходимый формат .avinput | |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -out refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ | аннотация snp (база данных refgene) | Основана на генной разметке |
| perl nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp_rs -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ | аннотация snp (база данных dbsnp)/ | Основана на фильтрации |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000g snp.avinput /nfs/srv/databases/annovar/humandb/ | аннотация snp (база данных 1000 genomes) | Основана на фильтрации |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ | аннотация snp (база данных Gwas) | Основана на разметке других регионов генома |
| perl /nfs/srv/databases/annovar/annotate_variation.pl snp.avinput /nfs/srv/databases/annovar/humandb/ -filter -dbtype clinvar_20140211 -buildver hg19 -out clinvar | аннотация snp (база данных Clinvar) | Основана на фильтрации |
> Разброс частот: cреднее значение частоты найденных snp - 0,35, минимум - 0,0034, максимум - 0,88.
Выводы из аннотации:
Таблицу со сводвой информацией о полиморфизмах можно скачать по ссылке: all_snp.xlsx Аннотация Refseq делит snp на три группы: "exonic" - 5, "intronic" - 12 и "3'-UTR" - 4. Проаннотированные снипы попали в три гена:
1. KCNJ11 - ген, кодирующий натриевый потенциал-зависимый канал (4 snp)
2. BUD13 - не удалось понять :( (10 snp)
3. ZPR1 - ген, кодирующий белок-энхансер сплайсинга пре-мРНК в моторных нейронах (7 snp)
2. BUD13 - не удалось понять :( (10 snp)
3. ZPR1 - ген, кодирующий белок-энхансер сплайсинга пре-мРНК в моторных нейронах (7 snp)
Также можно отметить, что из 21 snp rs имеют 19, данные по частотам приведены в таблице. Клиническая аннотация нашла только 2 snp. Про один из них не известно ничего кроме того, что он не является патогенным и неспецифичен, второй обладает такими же характеристиками, относится к диабету 2го типа.