Практикум 12
Сравните выравниваний разными программами
В задании по сравнению выравниваний я решила использовать последовательности с мнемоникой “CDD” (рекомендованное полное имя белка из ECOLI – цитидин деаминаза) из 9 практикума.
Для выполнения выравниваний я решила использовать программы Muscle , Mafft и T-Coffee. Полученные результаты можно посмотреть в проекте Jalview.
Попарные выравнивания белков: Muscle и Mafft, T-Coffee и Muscle
Для сравнения результатов выравнивания был использован сценарий Масленникова Вячеслава.
| Программы | Длина первого выравнивания | Процент совпадающих колонок | Длина второго выравнивания | Процент совпадающих колонок | Координаты блоков выравниваний |
|---|---|---|---|---|---|
| T-Coffe и Muscle | 314 | 67.2% | 313 | 67.41% | 15-20,45-55,66-97 103-130,133-136,140-153 169-170=170-171 175-191=176-192 195-288=196-289 313-314=312-313 |
| Mafft и Muscle | 315 | 76.19% | 313 | 76.68% | 28-32,36-130 134-153,158-164 176-283 |
Нетрудно заметить, что выравнивание T-Coffee меньше похоже на выравнивание Muscle, чем MAFFT. Если считать Muscle наиболее близким к "идеальному", то можно заключить, что программа T-Coffee работает хуже, чем MAFFT. Это может объясняться тем, что MAFFT вышла на 4 года позже, чем T-Coffee (2000 и 2004) и, видимо, использует менее совершенные алгоритмы.
Выравнивание по совмещению структур
Для анализа я выбрала семейство ABC-транспортеров, которые отвечают за перемещение различных соединений через биологические мембраны (PF00005).
Далее я нашла три белка, содержащие данный домен и имеющие структуры в PDB: 1GAJ, 1JI0, 1JJ7 (в качестве референсного выбрала белок человека 1JJ7).
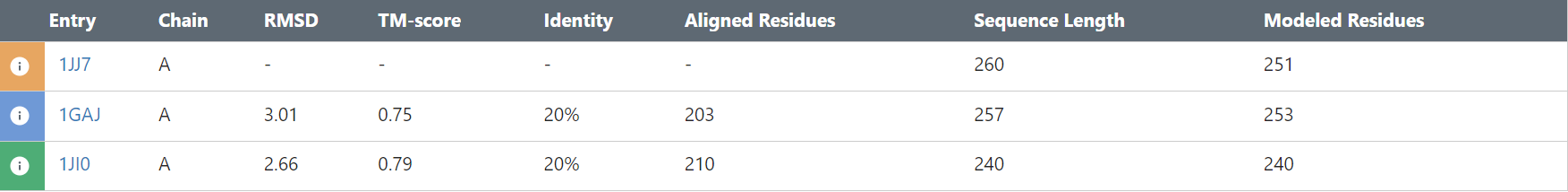
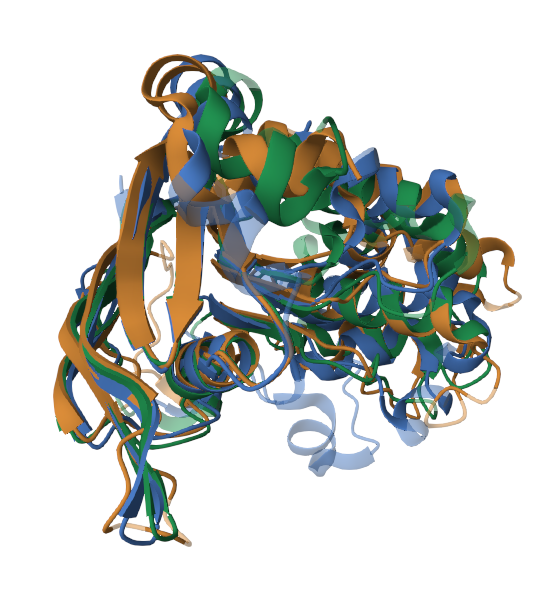
Затем было проведено попарное выравнивание структур (Рис.1). По алгоритму, описанному в подсказках было получено множественное выравнивание на основе совмещения структур. Последовательности тех же белков были выравнены с помощью программы Mafft.
На удивление выравнивания оказались достаточно схожими и имеют несколько одинаковых консервативных участков, что говорит о гомологичности белков.
Описание программы Muscle
Программа Muscle Alignment использует алгоритм MUSCLE, разработанный Робертом К. Эдгаром в 2004 году. Алгоритм состоит из трёх этапов:
1. Прогрессивный проект: на этом этапе алгоритм выполняет многократное выравнивание, уделяя внимание скорости, а не точности. Он начинается с вычисления расстояния k-мер для каждой пары входных последовательностей и создаёт матрицу расстояний. Затем UPGMA группирует матрицу расстояний, создавая двоичное дерево, на основе которого строится последовательное выравнивание.
2. Улучшенный прогрессивный: этот этап фокусируется на получении более оптимального дерева путём вычисления расстояния Кимуры для каждой пары входных последовательностей и создания второй матрицы расстояний. Затем UPGMA кластеризует эту матрицу, получая второе двоичное дерево. Прогрессивное выравнивание выполняется для получения оптимального выравнивания нескольких последовательностей.
3. Доработка: на этом заключительном этапе выбирается ребро из второго дерева, удаление которого разделяет дерево на два поддерева. Затем вычисляется профиль множественного выравнивания для каждого поддерева и создаётся новое выравнивание множественной последовательности путём повторного выравнивания профилей поддерева. Если оценка SP улучшена, новое выравнивание сохраняется, в противном случае оно отбрасывается. Процесс удаления ребра и выравнивания повторяется до сближения или пока пользователь не установит определённый предел.