Мне достался проект SRR4240356 по
секвенированию бактерии Buchnera aphidicola. В качестве подготовки необходимых файлов были скачаны
сами чтения, а также сохранены в рабочую папку и объединены в один файл остатки адаптеров командами:
Теперь, имея все необходимое для дальнейшей работы, стоит начать отбирать только самые лучшие чтения. Для
начала были удалены остатки адаптеров командой:
В полученном файле осталось 97.96% чтнений. Лог программы тут.
Теперь стоит удалить с правых концов чтений нуклеотиды с качеством ниже 20, оставьте только такие чтения,
длина которых не меньше 32 нуклеотидов. Я реализовал это командой:
В этот раз сохранились только 95.85% чтений файла без адаптера (305092 чтений было удалено на этом этапе).
Файл же стал меньше на 31,72 МБ (777308708Б было и 744045190Б стало).
Лог программы тут.
Подготовка k-меров
Теперь пора готовить k-меры для сборки, в данном случае их длина будет 31. Для этого использовалась
программа velveth после пары минут разбора параметров ее запуска:
velveth . 31 -short -fastq trimmed_reads.fatsq
Тут "." - команда на запись в текущую директорию, "31" - длина k-мера, "-short" сообщает о коротких
непарных чтениях во вносимом файле, "-fastq" показывает формат входного файла а "trimmed_reads.fatsq" - файл
с триммированными чтениями. Лог программы тут.
Результатом работы послужили три новых файла: Log, Roadmaps, Sequences.
Сборка
Пользуясь полученными данными, можно уже и начать сборку. Для этого использовалась программа velvetg:
velvetg . -cov_cutoff 30
Было выбрано значение "-cov_cutoff" 30 как, по идее, наиболее соответствующее самой идее графов де Брёйна -
длина k-мера минус один. Судя по логу
выполнения программы, N50 полученных контигов - 65554. Информация о трех самых длинных контигах находится в
таблице 1 и получена при помощи команды:
sort -n -r -k 2 stats.txt
Таблица 1. Три самых длинных контига
ID контига
7
5
9
Длина контига
111962
107488
80939
Покрытие контига
38.66
34.17
37.52
"Типичное" покрытие, ввиду того, что два контига длины один имеют покрытии по 266951 и 1134, что в разы
больше следующих за ними контигов (проанализировано по выдаче
Итак, медиана - 40,211. Очевидно, уже описанные контиги (их ID - 33 и 39) длины 1 и с зашкаливающими
порытиями очень сильно от нее отличаются. Впрочем, толькоо они - похоже, из-за своей ничтожной длины.
Анализ
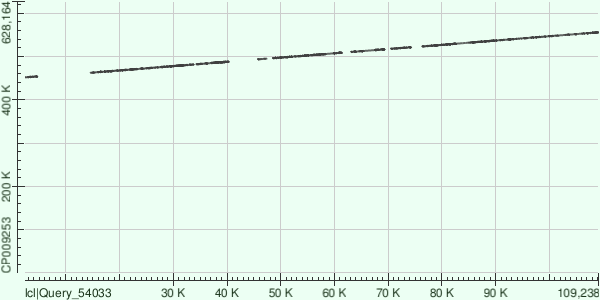
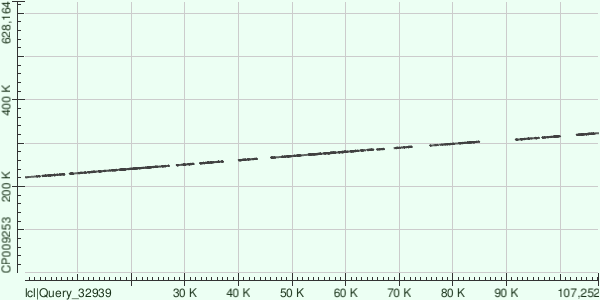
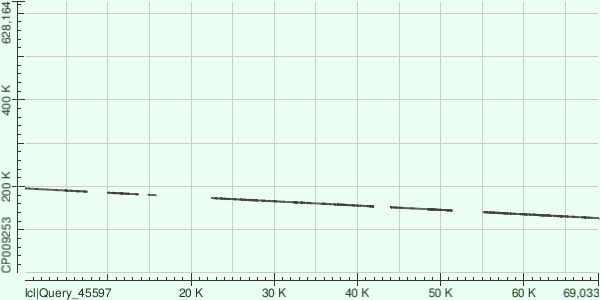
Далее каждый из трех самых длинных контигов (ID 7, 5, 9) был выровнен в megablast с уже имеющейся в
GenBank хромосомой Buchnera aphidicola
при стандартных настройках. Результатом этих выравниваний может служить таблица 2:
Сразу стоит отметить, что контиги наложились на известную последовательность далеко не по всей длине,
причем не выровнены куски не только с краев контигов - достаточно большие куски несоответствия находятся
и внутри последовательности контигов, так что в итоге покрытие во всех трех случаях не превышает трех
четвертей длины контига. Кроме того, в отличие от 5 и 7 контигов, 9 контиг наложился на референсный геном
в обратной последовательности, что можно объяснить не зависящиим от направления хромосомы секвенированием,
в результате которого и были получены исходные риды - часть прямых, часть обратных. В целом такой случай был
вполне ожидаем.
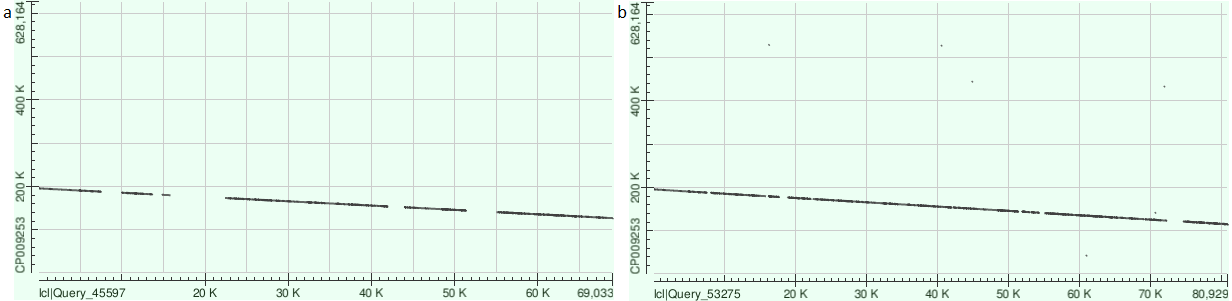
Что до довольно обширных зон несоответствия - мне не верится, что они действительно столь велики. Я
предположил, что такое их обилие вызвано эвристичностью алгоритма blast, и, чтобы проверить это, повторил
выравнивание, понизив длину слова до минимально возможной - 16. И да, на примере 9 контига пробелы между
участками заметно сократились (рис. 4), так что, похоже, при выравнивании, например, needle, в выдаче
окажутся хоть сколько-то длинные группы гэпов только в начале и конце, но такое выравнивание провести
было бы крайне ресурсозатратно. В общем, судя по хорошему выравнивнию, сборка произошла вполне успешно!
Рисунок 4. a - Dot plot контига 9 с длиной слова 28, b - Dot plot контига 9 с длиной слова 16.