Главная страница > Третий семестр > Структура тРНК
Рассмотрен pdb-файл 1n77, содержащий структуру глутамил-тРНК-синтетазы Thermus thermophilus, связанной с молекулами тРНКGlu, ATФ и ионом Mg2+. В файле представлены два таких комплекса, идентичных друг другу. Молекулы глутамил-тРНК-синтетазы были получены путем экспрессии соответствующих генов T.thermophilus в E.coli (в качестве вектора использована плазмида PK7), молекулы тРНКGlu - путем экспрессии генов T.thermophilus in vitro. Информация о молекулах и ионах, представленных в pdb-файле, приведена в табл. 1. Для дальнейшего исследования выбрана цепь C, соответствующая одной из тРНКGlu. Последовательность выбранной тРНК приведена ниже:
GGCCCCATCGTCTAGCGGTTAGGACGCGGCCCTCTCAAGGCCGAAACGGGGGTTCGATTCCCCCTGGGGTCACCA |
Данная тРНК включает в себя 77 нуклеотидов. Нумерация нуклеотидов начинается с номера 501. Так как максимальный номер аминокислотного остатка глутамил-тРНК-синтетазы - 468, то, скорее всего, нумерация нуклеотидов начата с 501 для того, чтобы их номера не совпадали с номерами аминокислотных остатков. Это делает работу со структурой в RasMol более удобной (например, команда RasMol select 508 выделяет только восьмой нуклеотид и не выделяет аминокислотных остатков). Номер последнего нуклеотида - 576. Имеется одна вставка в нумерации: один из уридиновых нуклеотидов имеет номер 520A. Рассматриваемая структура не содержит модифицированных нуклеотидов.
Табл. 1. Молекулы и ионы, представленные в pdb-файле 1n77.
|
Нуклеиновые кислоты |
||
| ID цепи |
|
|
| C |
|
|
| D |
|
|
|
Полипептидные цепи |
||
| ID цепи |
|
|
| A |
|
|
| B |
|
|
|
Другие молекулы и ионы |
||
| ID |
|
|
| MG |
|
|
| ATP |
|
|
| HOH |
|
|
Поиск пар азотистых оснований в структуре тРНК был проведен с помощью программы find_pair (см. выдачу программы при запуске с параметром -t). Так как структура тРНК значительно отличается от идеального дуплекса, то, возможно, имеет место образование водородных связей между более чем двумя остатками азотистых оснований. В связи с этим, программа find_pair была также запущена с параметрами -pt, что позволяет выявлять, помимо пар, тройки и более крупные ассоциации остатков азотистых оснований, а также водородные связи между нуклеотидными парами (однако, при запуске с данными параметрами в выдаче программы отсутствует информация о дуплексных участках, см. выдачу программы при запуске с параметрами -pt). Команды Linux, с помощью которых была запущена программа, приведены ниже:
find_pair -t
rna.pdb pairs.txt
find_pair -pt rna.pdb pairs2.txt
В составе молекулы тРНК find_pair выявила два дуплексных участка и одну изолированную нуклеотидную пару. Внутри каждого из дуплексов были выделены по два участка с непрерывной нумерацией обеих цепей. Таким образом, в состав структуры входят четыре спиральных участка (длиной от четырех до семи нуклеотидных пар). Ниже приведена последовательность тРНК, на которой нуклеотиды, входящие в состав различных спиральных участков, отмечены различными цветами. Каждый из выявленных спиральных участков соответствует одному из стеблей вторичной структуры тРНК: участок, выделенный красным, — акцепторному, синим — антикодоновому, зеленым и оранжевым — DU- и TψC-стеблям.
GGCCCCA TC GTCT AGCGGTTA GGAC GCGGCCC TCTCA AGGCCGA AAC GGGGG TTGATT CCCCC TGGGGTC ACCA |
Из 28 выявленных нуклеотидных пар 9 оказались неканоническими (например, GU, AA или AU с отличным от канонического расположением водородных связей). Присутствие большого количества неканонических пар может быть объяснено тем, что, в отличие от ДНК, молекулы РНК не находятся под контролем системы репарации. Таким образом, образуются такие нуклеотидные пары, которые минимизируют суммарную энергию молекулы. В отличие от РНК, в ДНК существуют только те пары, которые не исправляются системой репарации.
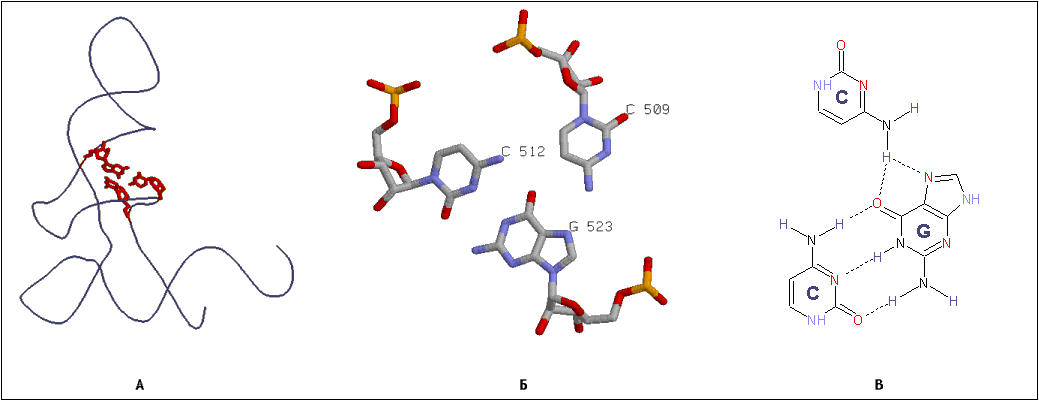
Помимо пар остатков азотистых оснований, программой find_pair было выявлено 5-7 более сложных структур, состоящих из нескольких остатков азотистых оснований, стабилизируемых водородными связями (например, тройка С509-С512-G523, см. выдачу программы). Предположительная структура водородных связей внутри этой тройки показана на рис. 1. Функция таких структур может заключаться в дополнительной стабилизации третичной структуры тРНК.
Рис. 1 (А, Б, В). Тройка азотистых оснований С509-С512-G523, выявленная в составе тРНК с помощью программы find_pair. А. Расположение тройки в третичной структуре молекулы (нуклеотиды тройки показаны красным). Б. Структура тройки (изображение получено с помощью RasMol). В. Водородные связи, предположительно образуемые остатками азотистых оснований тройки друг с другом.
Трехмерная структура тРНК в остовной модели представлена на рис. 2 (отмечены дуплексные участки, выявленные с помощью find_pair). Скрипт, генерирующий данное изображение, приведен здесь.
В составе тРНК выявлены три участка внеспиральных стекинг-взаимодействий. Один из участков состоит из четырех нуклеотидов, находится на антикодоновой петле и, скорее всего, стабилизирует ее структуру (рис. 3А). Возможно, это повышает точность узнавания кодонов мРНК. Второй участок располагается в центре структуры и включает в себя семь нуклеотидов (рис. 3Б). Данный участок может играть важную роль при формировании третичной структуры тРНК, так как он стабилизирует сближенные нуклеотиды из отдаленных участков цепи. Третий участок включает в себя три нуклеотида из одноцепочечного участка акцепторного стебля (3'-конец молекулы, рис. 3В). Стабилизация данного участка, возможно, необходима для его узнавания аминоацил-тРНК-синтетазой. Скрипт, с помощью которого могут быть получены изображения внеспиральных стекинг-взаимодействий, приведен здесь.
С помощью программы find_pair были выявлены 9 неканонических пар в составе тРНК (см. выше). Структура водородных связей между азотистыми основаниями пар GU, AA и AU представлены на рис. 4 (водородные связи показаны в соответствии с выдачей программы find_pair при запуске с параметрами -pt).
Принадлежность структуры тРНК к A-форме нуклеиновых кислот определена на примере дуплексного участка, выделенного красным на рис. 2. Данный участок был сохранен в отдельном файле с помощью следующих команд RasMol:
script rna.txt
restrict helix_1
save pdb helix1.pdb
Структура данного участка соответствует структуре A-формы нуклеиновых кислот (см. "A- и B-формы ДНК"):
глубина большого желобка (желобка, в который обращены аминогруппы остатков аденина канонических пар) превышает радиус дуплекса, что приводит к тому, что в центре проекции структуры располагается отверстие;
плоскости азотистых оснований расположены под углом около 71° к оси молекулы;
атомы кислорода фосфатов обращены внутрь большого желобка;
ширина большого желобка (9.1 Å, измерена от 501-го нуклеотида) меньше ширины малого желобка (15.4 Å, измерена от 506-го нуклеотида), оба значения согласуются с соответствующими значениями для A-формы НК;
по результатам analyze, (1) подавляющее большинство остатков рибозы расположены в C3'-эндо-конформации, что характерно для A-формы НК, (2) торсионные углы и некоторые другие параметры структуры (значения, характеризующие отклонения остатков азотистых оснований от идеального расположения) близки к соответствующим параметрам A-формы НК.
Принадлежность структуры тРНК к A-форме может быть объяснена воздействием на конформацию молекулы 2'-OH-групп рибозы [1].
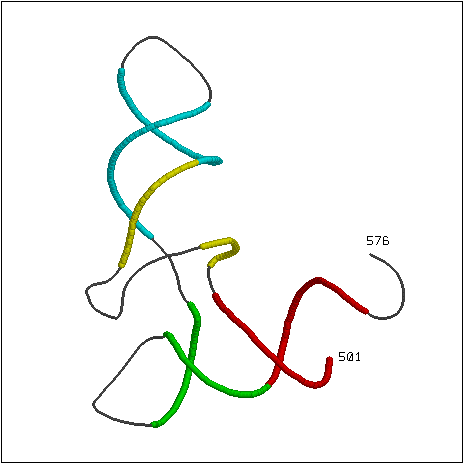
Рис. 2. Дуплексные участки в составе молекулы тРНК. Нуклеотиды дуплексных участков выделены цветами, указаны номера первого и последнего нуклеотидов.
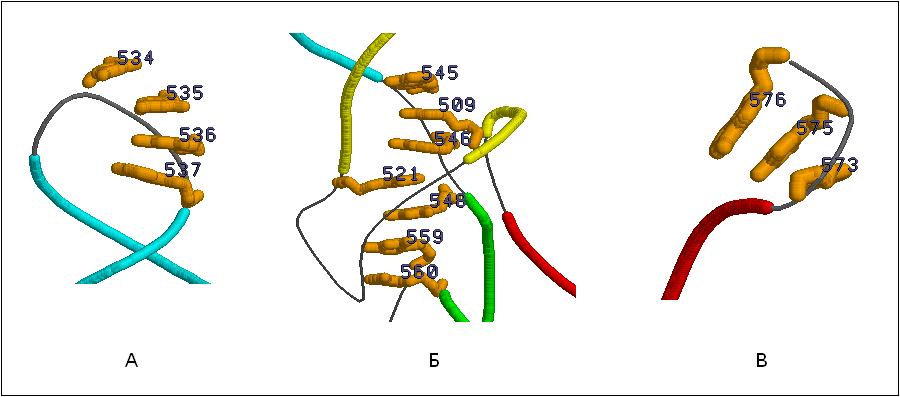
Рис. 3 (А, Б, В). Внеспиральные стекинг-взаимодействия в молекуле тРНК (А, Б, В - различные участки таких взаимодействий, комментарии см. в тексте). Дуплексные участки тРНК выделены цветом.
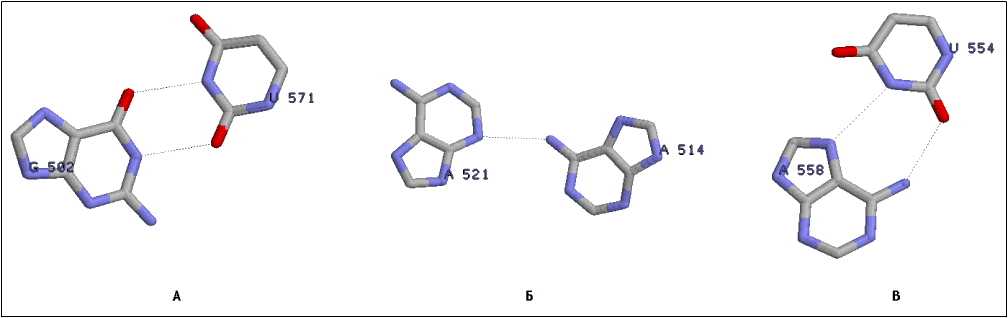
Рис. 4 (А, Б, В). Структура водородных связей внутри некоторых неканонических пар азотистых оснований (А - внутри пары GU, Б - внутри пары AA, В - внутри пары AU с отличной от канонической структурой водородных связей)
С помощью программы einverted был проведен поиск участков инвертированного сходства в составе последовательности тРНК. Поиск проведен с различными значениями параметров -gap (штраф за гэп), -match (баллы, начисляемые за канонические пары), -mismatch (баллы, начисляемые за неканонические пары); значение параметра -threshold (пороговое значение) принято равным пяти. Пример команды Linux, с помощью которой была запущена программа, приведен ниже:
einverted trnaseq.fasta -gap 5 -threshold 5 -match 5 -mismatch 0 result.txt result.fasta
Ни при одной из комбинаций значений параметров программы результат не совпал с данными рентгеноструктурного анализа (РСА), представленным в pdb-файле. Выдача программы при запуске с параметрами, указанными в командной строке, приведена здесь. Помимо двух участков, состоящих из нескольких комплементарных нуклеотидов (GC-пар), входящих в состав акцепторного и антикодонового стеблей, ни один из выявленных участков инвертированного сходства не совпал с реальными дуплексными участками.
Сложности, связанные с предсказанием вторичной структуры с помощью einverted, обусловлены двумя причинами.
В составе тРНК имеется большое количество неканонических пар нуклеотидов. Программа einverted проводит поиск участков инвертированного сходства основываясь только на канонических взаимодействиях между нуклеотидами. Это затрудняет выявление реальных дуплексных участков.
Фрагменты последовательности тРНК, участвующие в образовании дуплексов, имеет длину, не превышающую семь нуклеотидов. В результате вероятность обнаружения участков инвертированного сходства той же длины и с тем же процентом комплементарных пар, не соответствующих реальным дуплексным участкам, оказывается достаточно высокой.
Следует отметить, что успешно выявляемые с помощью einverted реальные дуплексные участки состоят преимущественно из GC-пар. Это связано с тем, что пара нуклеотидов GC наиболее прочная по сравнению с другими каноническими и неканоническими парами (между азотистыми основаниями образуется три водородных связи). Таким образом, при сворачивании тРНК преимущественно происходит образование пар между остатками гуанинов и цитозинов, чем между остатками гуанинов и других оснований и остатков цитозинов и других оснований. Так как пара GC является канонической, она повышает баллы участков инвертированного сходства, выявляемых программой einverted. Следовательно, если в составе последовательности тРНК присутствуют два комплементарных участка, состоящих из остатков G и C, то они, скорее всего, образуют дуплекс и будут выявлены с помощью einverted.
Предсказание вторичной структуры тРНК также проведено с помощью программы mfold. Запуск программы осуществлен с параметром P, равным 20 (для данной тРНК программа выдала три наилучших предсказания). Команда Linux, с помощью которой была запущена программа, приведена ниже:
mfold SEQ=trnaseq.fasta P=20
Были построены три варианта вторичной структуры с энергиями Гиббса -36.70, -31.51 и -27.41 кДж/моль. Вариант вторичной структуры с наименьшей энергией (рис. 5) в целом соответствует реальной структуре тРНК. В выдаче программы присутствуют все элементы вторичной структуры, характерные для тРНК: акцепторный стебель, TψC- и DU-петли и стебли, антикодоновая петля и антикодоновый стебель, вариабельная петля.
Расхождения заключаются в отличии некоторых предсказанных с помощью mfold пар, располагающихся на концах стеблей, от реальных. Это может быть связано с тем, что нуклеотиды, располагающиеся на концах дуплексов, испытывают более разнообразные воздействия со стороны других нуклеотидов, чем только стекинг и водородные связи между парами комплементарных оснований (например, спаривание троек оснований, водородные связи, образуемые основаниями с атомами остатков дезоксирибозы и кислородами фосфатов, внеспиральный стекинг). Эти взаимодействия не предусмотрены алгоритмом программы mfold и, следовательно, не могут быть успешно предсказаны.
В целом, предсказание mfold согласуется данными РСА в отличие от предсказаний einverted. Это связано с тем, что mfold учитывает вклад не только взаимодействий между комплементарными парами азотистых оснований, но и взаимодействий между некомплементарными парами, а также стекинг-взаимодействия. Результаты применения mfold показали, что данная программа может быть успешно использована для предсказания вторичной структуры молекул РНК, длина которых составляет 50-100 н.п.
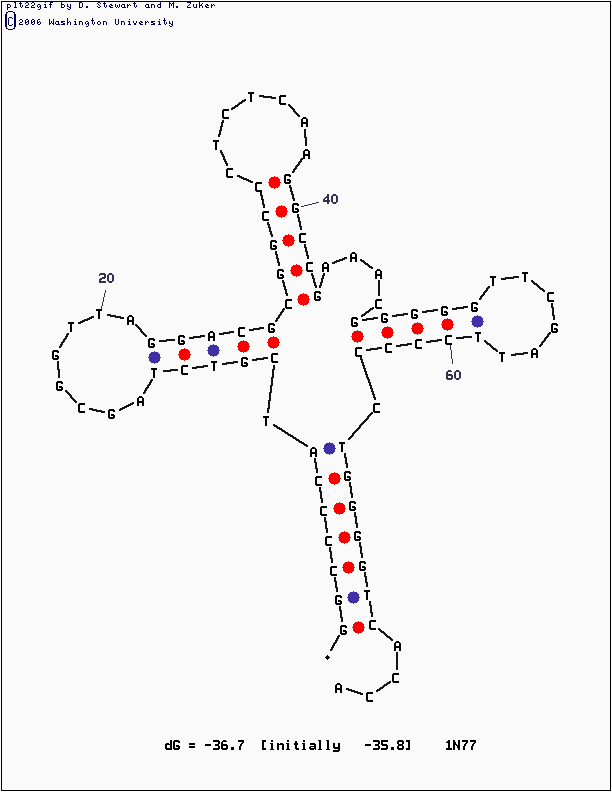
Рис. 5. Наилучший вариант вторичной структуры тРНК, предсказанный с помощью программы mfold (ΔG = -36.7 кДж/моль).
1. Lodish, Berk, Zipursky, Matsudaira, Baltimore and Darnell Molecular Cell Biology, 4th edition. New York: W. H. Freeman & Co.; 2000.
© Куравский Михаил Львович, 2006