Multiple alignment of protein sequences
Comparison of alignment of the same sequences by different programs
I chose 25 enolase sequences from Swiss-Prot for comparison using Crustal Omega and CrustalW. So the results of MSA can be found via the links: for the CrustalW and for the Crustal Omega. For comprasion I'd used compare_alig program (made by E. Pleshko). Output table is on a separate page.
It's clear that the most of the alignment columns have coincided, with the exceptions being only a few: 148-150, 171-176, 212-218 and another. The proportion of equally aligned positions in both alignments is 0.92%. In conclusion the alignments of these two programs practically don't differ which is logical since both programs are from one series of program — Clustal.
Structural alignment using PyMol
I made a structural alignment using align command in PyMol. For the alignment I used three PDB structures predicted by AlphaFold (taken from Swiss-Prot in UniProt): CRISPR system endoribonuclease Csm6, CRISPR type III-A/MTUBE-associated protein Csm6, endopeptidase La from PF09659 Pfam family which is named CRISPR-associated protein (Cas_Csm6).
As we can see the structures didn't coincide by 100%, but homology in individual elements can be observed (in the β-sheets and in the two central α-helixes).
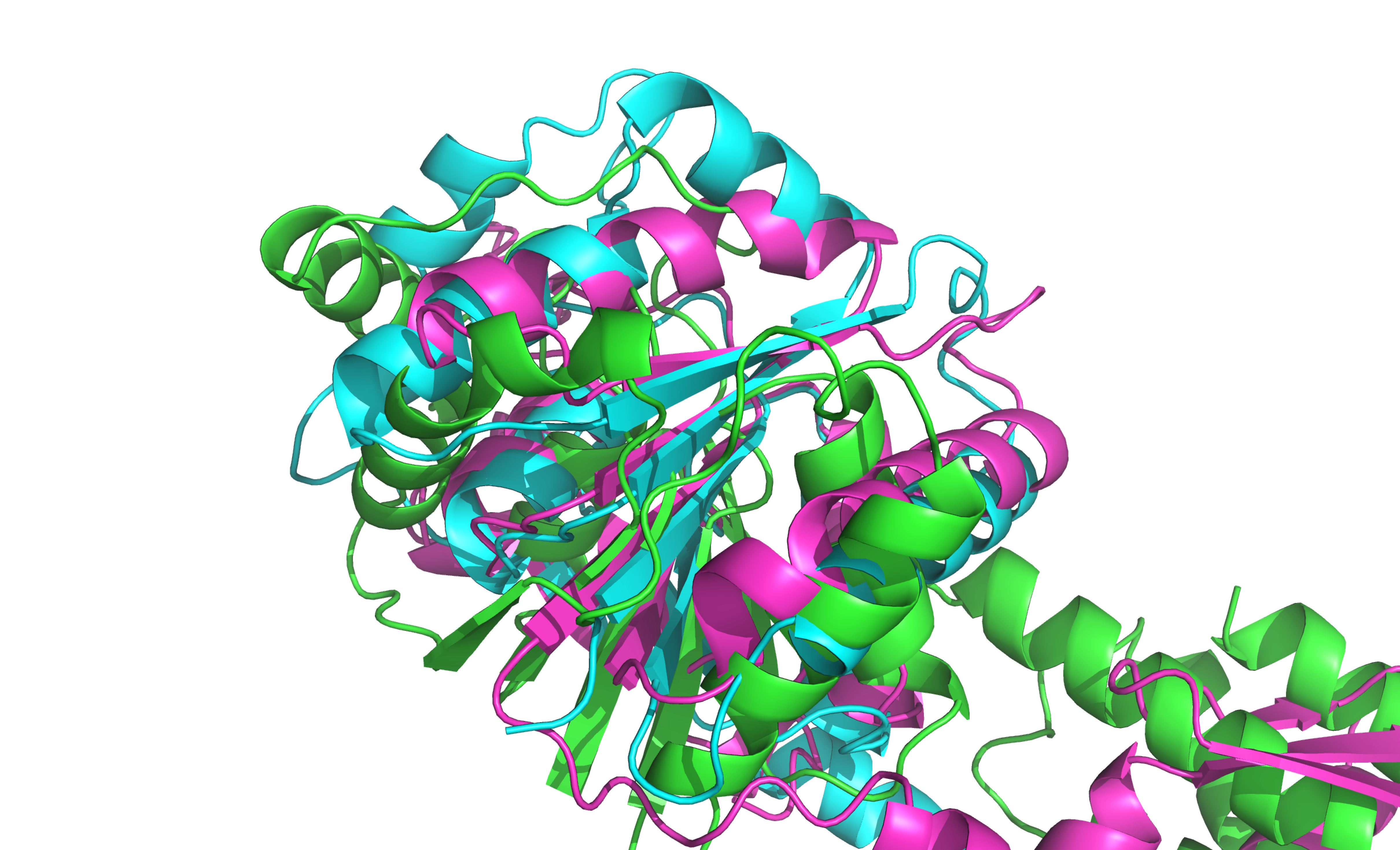
Fig. 1. Structural alignment of three proteins from PF09659 Pfam family.
Clustal Omega description
Clustal Omega (ClustalΩ, Clustal O) is the current standard version of the Clustal software used for multiple sequence alignment. There are five main steps.
- The first is pairwise alignment using the k-tuple method.
- After that the sequences are clustered using the modified mBed method which calculate pairwise distance using sequence embedding.
- Than the k-means clustering method is used.
- Next, the guide tree is constructed using the UPGMA method.
- The MSA is produced using HHAlign package from the HH-Suite, which uses two profile HMM's. A profile HMM is a linear state machine consisting of a series of nodes, each of which corresponds roughly to a column in the alignment.
The accuracy of Clustal Omega on a small number of sequences is very similar to what are considered high quality sequence aligners. The difference comes when using large sets of data with hundreds of thousands of sequences when Clustal Omega outperforms other algorithms across the board.
References
Pais F.S., Ruy P.C., Oliveira G., Coimbra R.S. (2014). Assessing the efficiency of multiple sequence alignment programs. Algorithms for Molecular Biology. 9(1): 4. doi:10.1186/1748-7188-9-4. PMC 4015676. PMID 24602402.
Blackshields G., Sievers F., Shi W., Wilm A., Higgins D.G. (2010). Sequence embedding for fast construction of guide trees for multiple sequence alignment. Algorithms for Molecular Biology. 5: 21. doi:10.1186/1748-7188-5-21. PMC 2893182. PMID 20470396.
Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J.D., Higgins D.G. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 7(1): 539. doi:10.1038/msb.2011.75. PMC 3261699. PMID 21988835.
Daugelaite J., O'Driscoll A., Sleator R.D. (2013). An Overview of Multiple Sequence Alignments and Cloud Computing in Bioinformatics. ISRN Biomathematics. 2013: 1–14. doi:10.1155/2013/615630. ISSN 2090-7702.