|
|
|
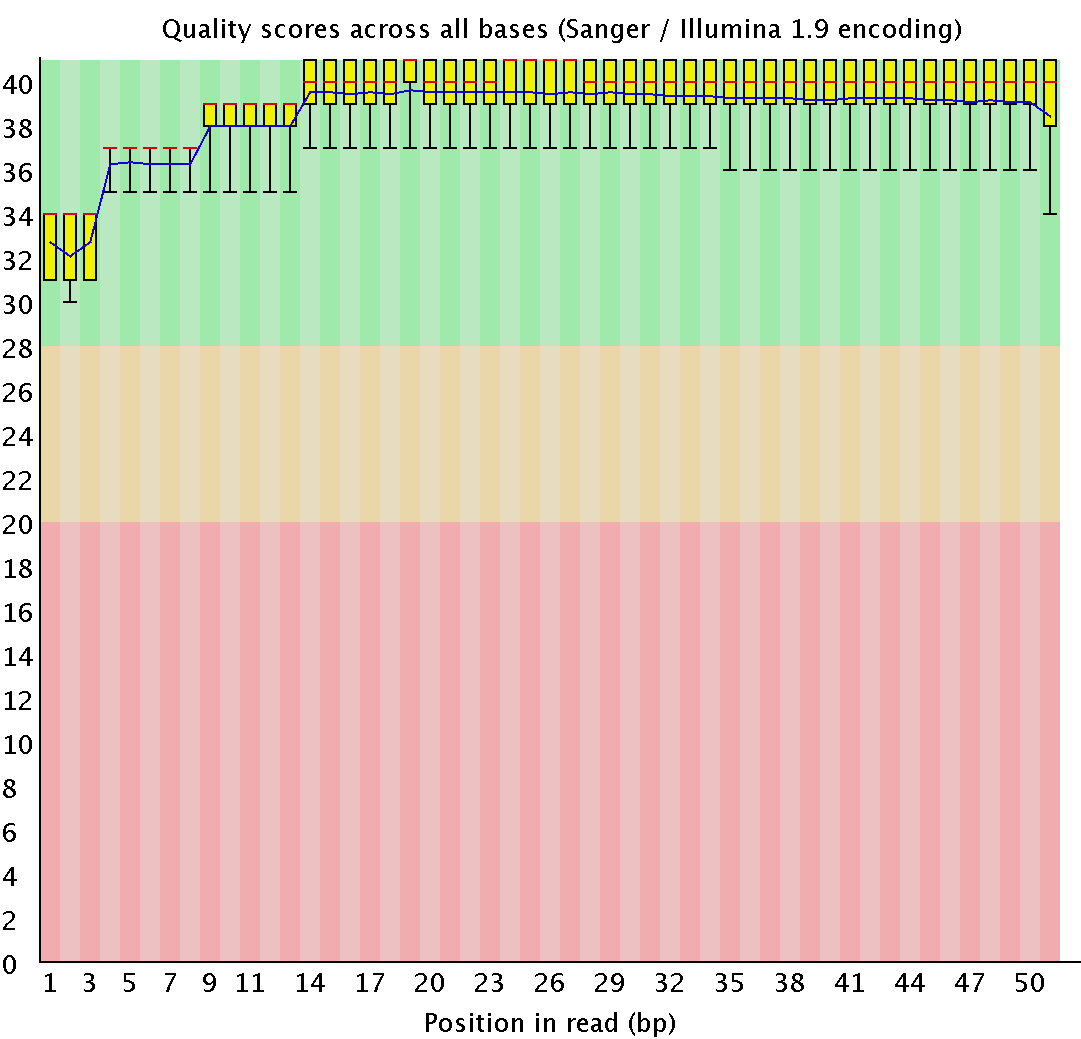
- Task 1: FASTQC usage for .fastq file analyse: command - fastqc chr7.2.fastq. Result is presented below:
|
|
|
- Task 2: There was no need to use Trimmomatic to cut off end low quality (<20) nucleotides and leave >50 length reads, because of high original quality. Amount of reads - 13701.
|
|
| fastqc chr7.fastq |
File analyse with FASTQC programm |
| hisat2-build chr7.fasta chr7 |
Indexes reference sequence |
| hisat2 --no-softclip -x chr7_neue -U chr7.2.fastq -S alignment1.sam |
Alignment of the reference sequence |
| samtools view alignment1.sam -b -o alignment1.bam |
SAM -> BAM (binary) |
| samtools sort alignment1.bam -T alignment1.txt -o samsorted1.bam |
Sorts alignment file; writes temporary files to PREFIX.nnnn.bam |
| samtools index samsorted1.bam |
Indexing of samsorted.bam |
|
- Task 3: Mapping:
- --no-spliced-alignment was excluded because this parameter is unnesessary for transcriptome. After hisat2 commands had been runned 8 files were created, according to them: 122 reads were not aligned, 13529 were aligned once and 50 were aligned more times.
|
|
|
- Task 4: Alignment analysis:
- Used commands:
- samtools view alignment1.sam -b -o alignment1.bam
- samtools sort alignment1.bam -T alignment1.txt -o samsorted1.bam
- samtools index samsorted1.bam
- Output file contains different information: read name, name of the reference sequence, number of mismatches in the alignment, number of mapped locations for the read or the pair, mapping quality, coordinates of the read's alignment beginning on the reference sequence etc.
|
|
|
- Task 5: Bedtools:
- Commands:
- bedtools bamtobed -i samsorted1.bam > samsorted1.bed (converting bam to bed)
- bedtools intersect -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b samsorted1.bed -u > final.bed (coincidence search between two sets of genomic features)
- Results:
- CPED1 - Cadherin like and PC-esterase domain containing 1 coding gene, locates in ER. Its function is unknown. The biggest part of reads belongs exactly to this gene - 219.
- 3 pseudogenes: HMGN1P18, RNA5SP241 and RNU6-517P. Each one of them got a little part of reads - 3.
|
|
|
- Pr 13. Bedtools. Additional task:
- Commands:
- Receive file with reads from the file with alignment: bedtools bamtofastq -i alignment1.bam -fq alignment1.fq
- Combine reads into clusters: bedtools cluster -i samsorted1.bed -d 5 > cluster1.txt
- Divide your chromosome into 1 million lenth fragments: bedtools makewindows -g chr7.txt -w 1000000 > div.txt. Chromosomes lenth - 159138663, number of fragments - 160.
|
|
|
|