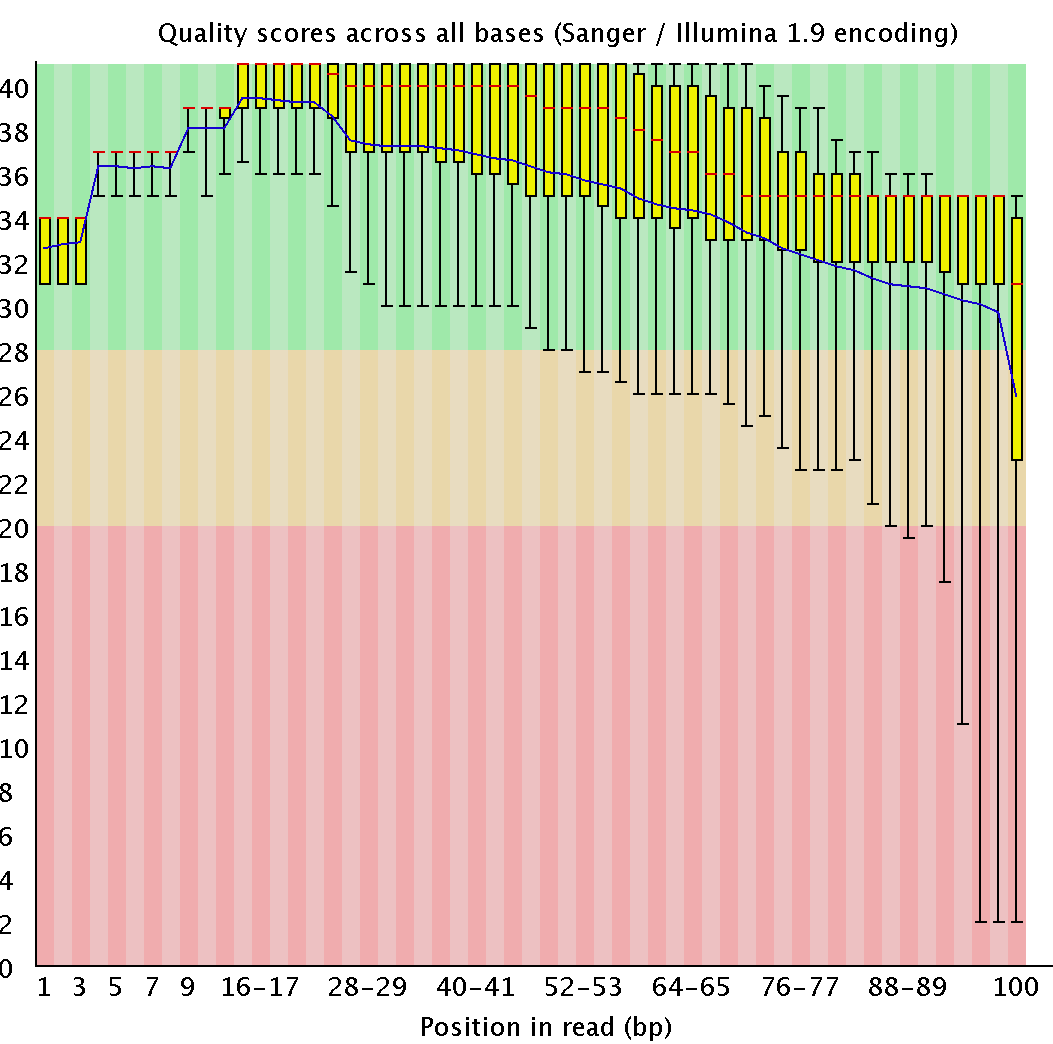
| fastqc chr7.fastq |
File analyse with FASTQC programm |
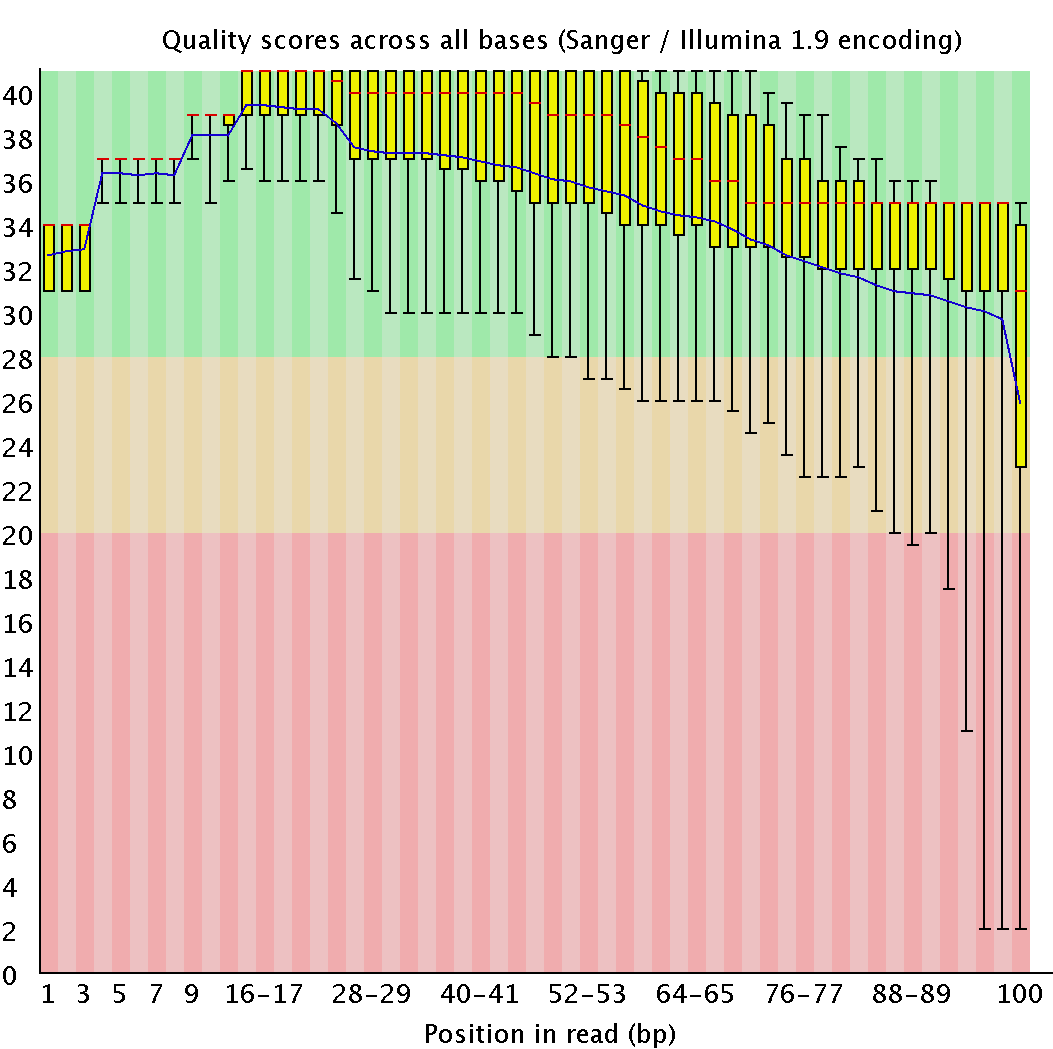
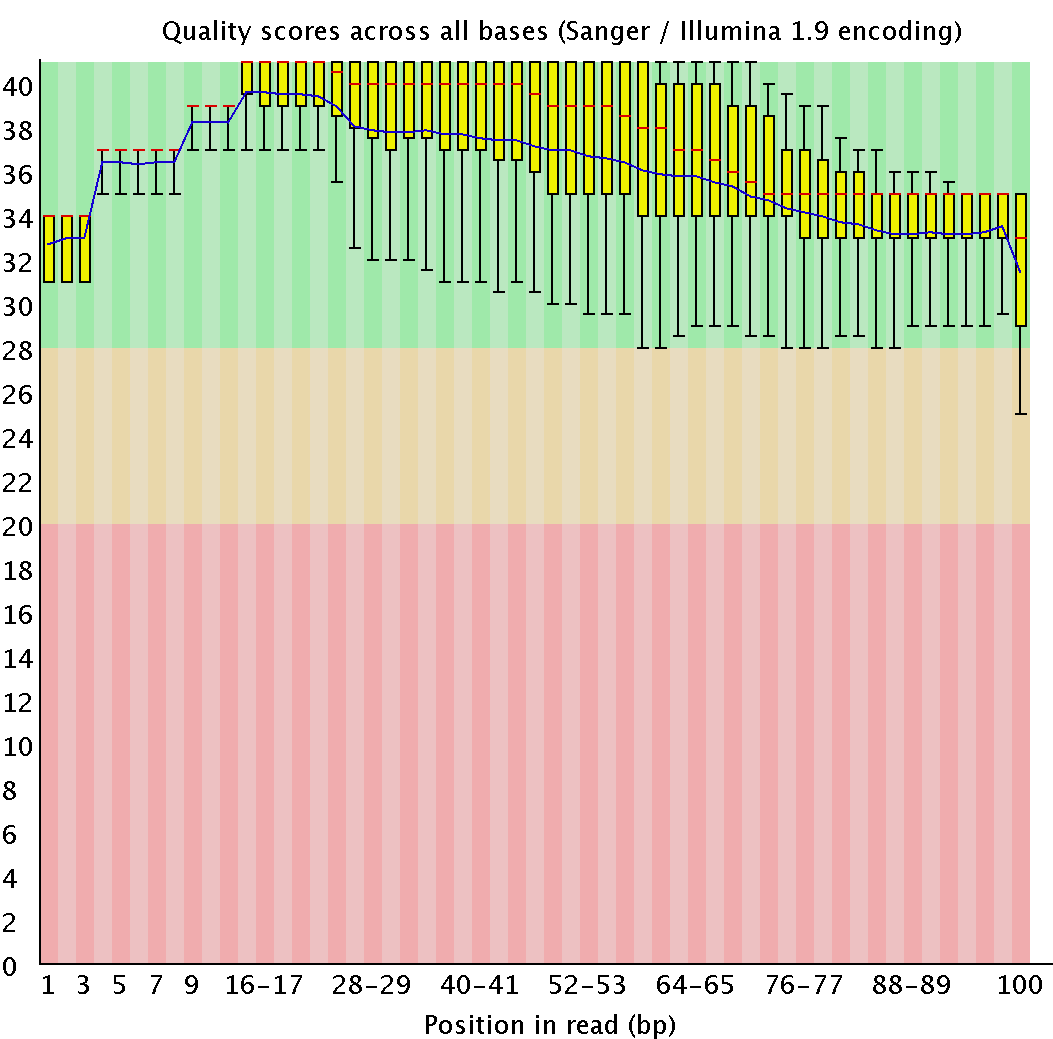
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr7.fastq chr7.1.fastq TRAILING:20 MINLEN:50 |
Cut off end low quality (<20) nucleotides; >50 length reads are left |
| hisat2-build chr7.fasta chr7his |
Indexes reference sequence |
| hisat2 --no-spliced-alignment --no-softclip -x chr7his -U chr7.1.fastq -S alignment.sam |
Alignment of the reference sequence |
| samtools view alignment.sam -b -o alignment.bam |
SAM -> BAM (binary) |
| samtools sort alignment.bam -T alignment.txt -o samsorted.bam |
Sorts alignment file; writes temporary files to PREFIX.nnnn.bam |
| samtools index samsorted.bam |
Indexes samsorted.bam |
| samtools mpileup -uf chr7.fasta samsorted.bam -o snp.bcf |
Creates .bcf file (with polymorphisms) |
| bcftools call -cv snp.bcf -o SNP.vcf |
Creates file with diefferences between reference and read in .vcf format |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_vcf.vcf > chr7.avinput |
Creates file.avinput |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr7.snp -build hg19 -dbtype snp138 chr7.avinput /nfs/srv/databases/annovar/humandb/ |
SNPs with rs |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -out refgene -build hg19 chr7.avinput /nfs/srv/databases/annovar/humandb/ |
refGene annotation |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out dbsnp -build hg19 -dbtype snp138 chr7.avinput /nfs/srv/databases/annovar/humandb/ |
dbsnp annotation |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000g chr7.avinput /nfs/srv/databases/annovar/humandb/ |
1000 genomes annotation |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out GWAS -dbtype gwasCatalog chr7.avinput /nfs/srv/databases/annovar/humandb/ |
GWAS annotation |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out clinvar -dbtype clinvar_20150629 -buildver hg19 chr7.avinput /nfs/srv/databases/annovar/humandb/ |
Clinvar annotation |