
A- и В- формы ДНК. Структура РНК
- Содержание:
- Средства JMol для работы со структурами нуклеиновых кислот
- Сравнение структур 3-х форм ДНК с помощью средств JMol
- Определение параметров структур нуклеиновых кислот с помощью программ пакета 3DNA
- Исследование ДНК-белковых взаимодействий в структуре комплекса белка и фрагмента ДНК
- Исследование структуры тРНК
Построение модели структур A-, B- и Z-формы ДНК с помощью инструментов пакета 3DNA
Пакет 3DNA один из популярных пакетов программ для анализа и простейшего моделирования структур нуклеиновых кислот.
Работает под операционной системой LINUX.
С помощью программы Рutty, используя протокол ssh, подсоединяемся к серверу kodomo.cmm.msu.ru.
Переходим в директорию term3/block1/pr2. Вводим следующие команды, что бы указать путь к 3DNA:
export X3DNA=/home/preps/golovin/progs/X3DNA
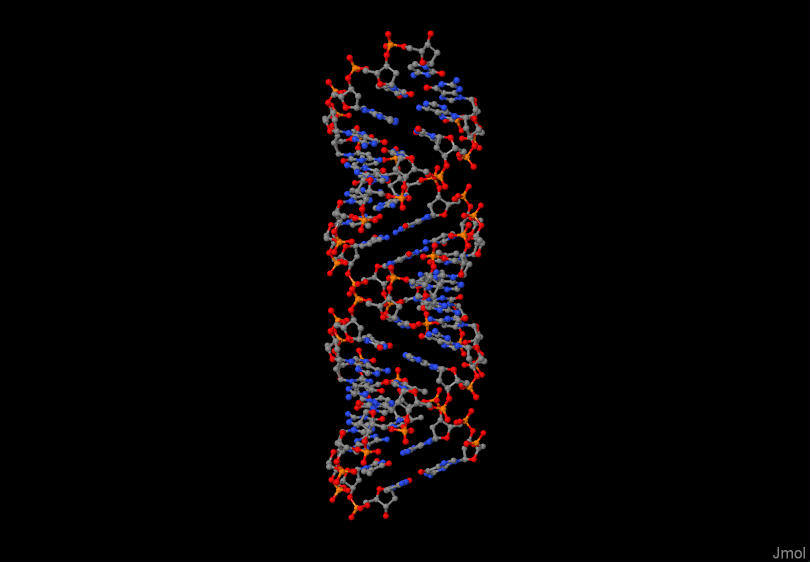
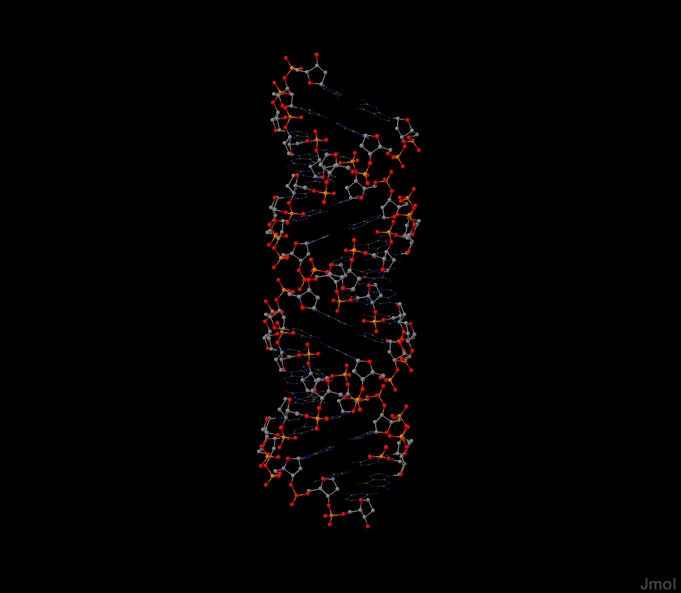
С помощью программы fiber пакета 3DNA строим A-, B- и Z-форму дуплекса ДНК, последовательность одной из нитей которого представляет собой 5 раз повторенную последовательность "gatc". Для этого используем команды :
fiber -b gatc-b.pdb
fiber -z gatc-z.pdb
Структура дуплекса в А-форме находится в файле gatc-a.pdb, структура дуплекса в В-форме в файле gatc-b.pdb, структура дуплекса в Z-форме в файле gatc-z.pdb.
| А-форма | B-форма | Z-форма |
 |
 |
 |
 |
 |
 |
Работа со структурами нуклеиновых кислот с помощью средств программы JMol
Используя файл gatc-a.pdb для А-формы, выделим цветом или способом отображения:
- сахарофосфатный остов ДНК;
- все нуклеотиды;
- все аденины;
- атом N7 во всех гуанинах и/или только в первом по последовательности.
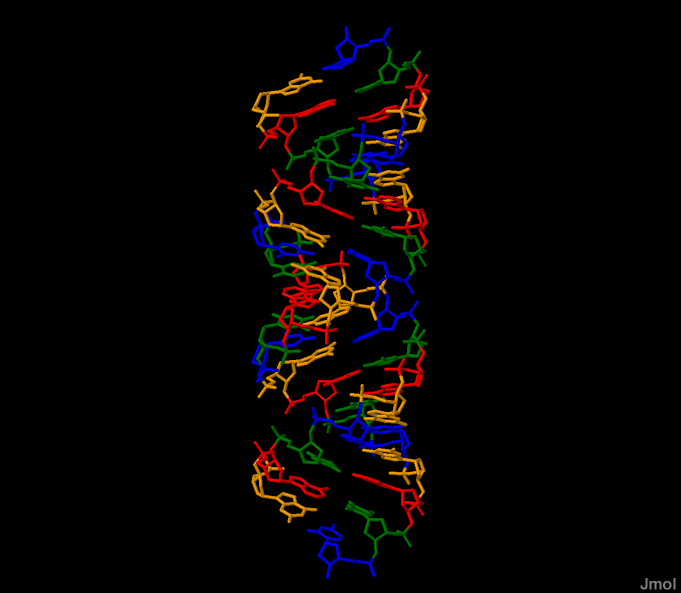
Нуклкотиды выделены в соответсвии с данной цветовой гаммой:
Аденин - красный, тимин - зеленый, гуанин - оранжевый, цитозин - синий.
Получение файлов .pdb по идентификаторам 1G59 и 1DDN
На сайте PDB выполним поиск по идентификаторам 1G59 и 1DDN. Выберем подходящие варианты структур.
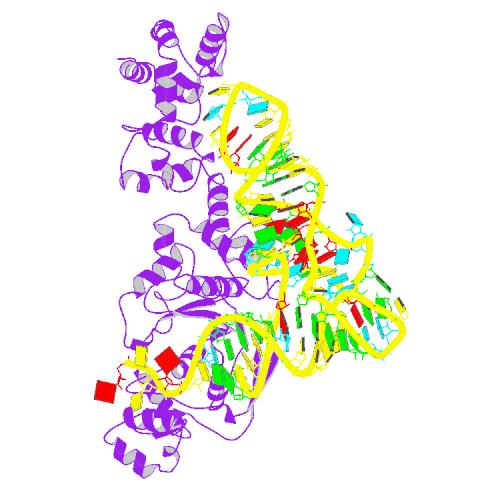
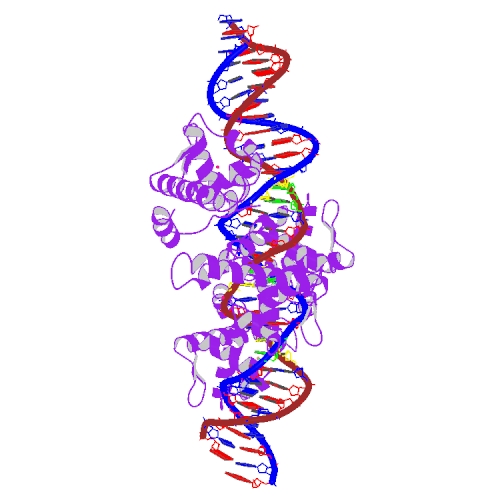
Выбранные варианты структур, представленные выше, сохранены в файлах 1G59.pdb и 1DDN.pdb соответственно.
Проверка заданных структур ДНК и РНК на наличие разрывов
Структуры 1DDN и 1G59 можно рассмотреть в программе JMol, изучив их на наличие разрывов. В них разрывы отсутствуют. Ниже представлены изображения только нуклеиновой кислоты в проволочной модели для рассматриваемых структур.
На рисунке представлена D-цепь РНК из файла pdb.

В файлах 1DNN_DNA.txt и 1G59_RNA.txt сохранены координаты атомов только РНК и ДНК соответственно.
Сравнение структур 3-х форм ДНК с помощью средств JMol
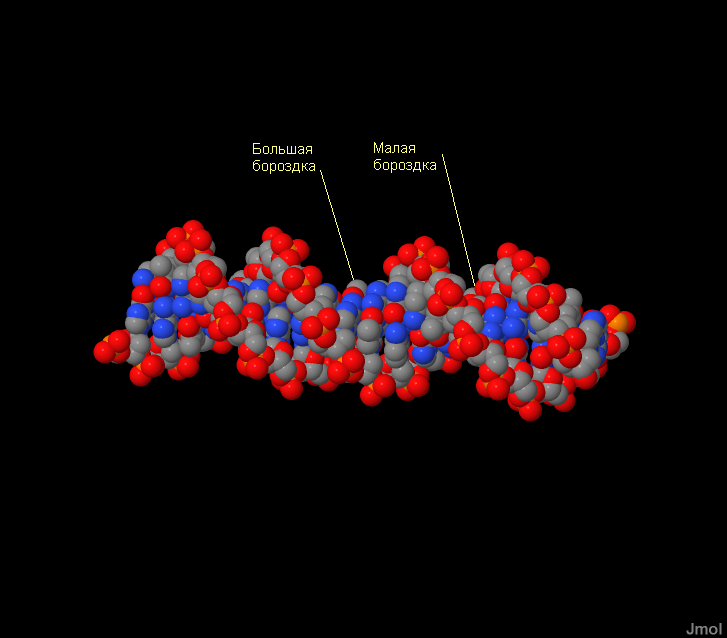
Большие и малые бороздки
Научимся находить большие и малые бороздки. Откроем в JMol файл gatc-b.pdb, полученный ранее. Рассмотрите структуру и визуально определим большую и малую бороздку (большая бороздка не обязательно шире малой, но обязательно глубже).
На рисунке виделены большая и малая бороздки.
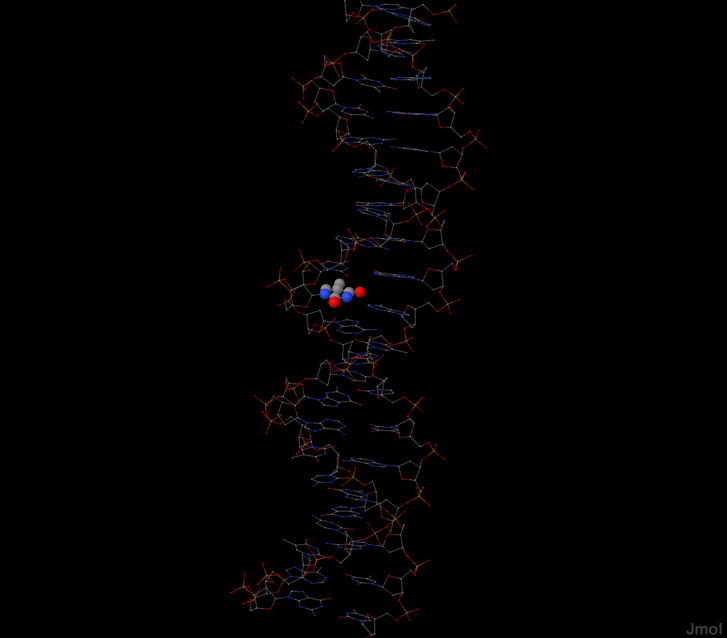
Теперь выберем теперь тимин в любом удобном для нас месте структуры, наприме 11 Тимин.
Определим, какие атомы основания явно обращены в сторону большой бороздки,
а какие в сторону малой.
С помощью ChemSketch получим изображение основания, выделим красным цветом атомы, смотрящие в сторону большой бороздки, синим - в сторону малой.
Красным цветом отмечены атомы, смотрящие в сторону большой бороздки,
синим - в сторону малой.
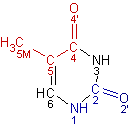
В сторону большой бороздки обращены атомы: T11.С4, T11.С5, T11.O4', T11.С5M
В сторону малой бороздки обращены атомы: T11.С2, T11.N1, T11.O2'
Остальные атомы основания: T11.С6, T11.N3
В Z-форме нет тиминов, а в A-форме атомы тимина обращены с точностью в противоположенные стороны с атомами в В-форме:
В сторону большой бороздки обращены атомы: T11.С2, T11.N1, T11.O2'
В сторону малой бороздки обращены атомы: T11.С4, T11.С5, T11.O4', T11.С5M
Сравнение основных спиральных параметров разных форм ДНК
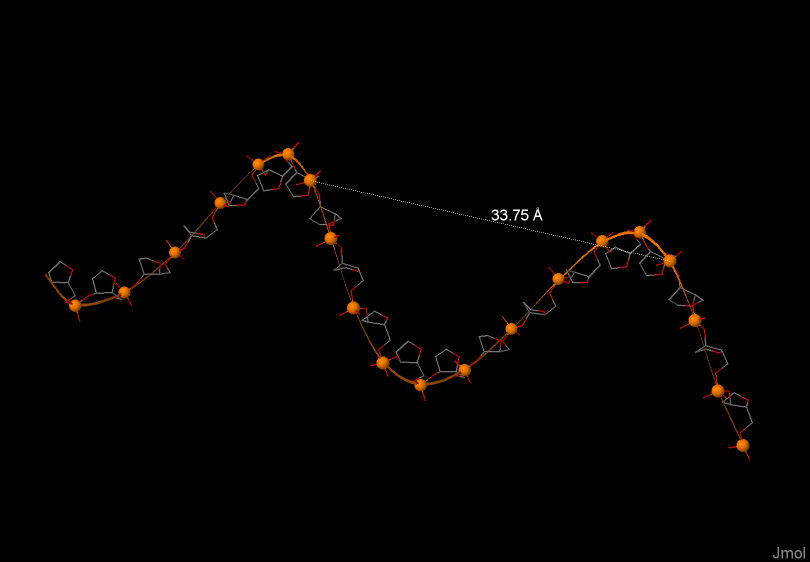
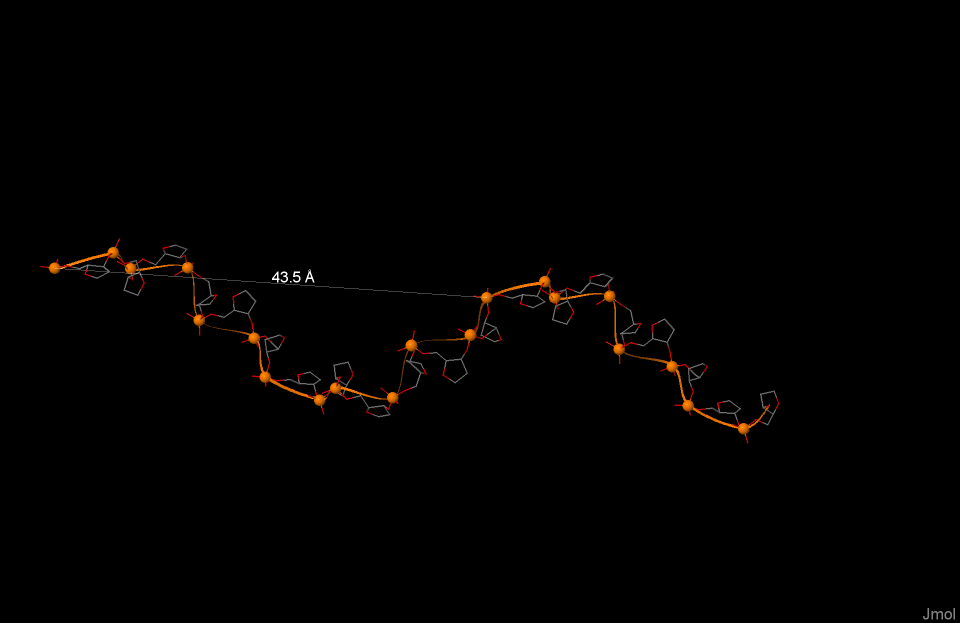
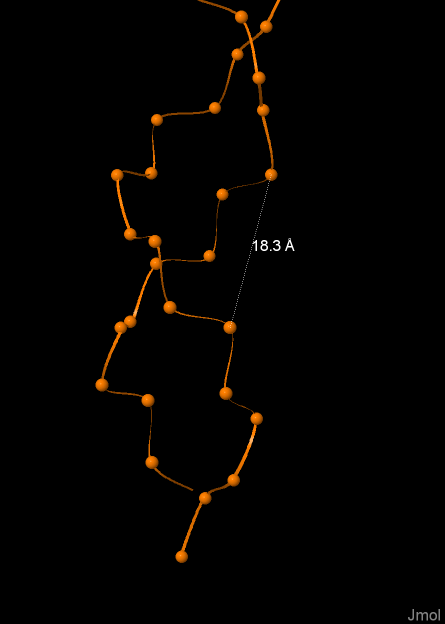
На рисунах 4, 5, 6 изображена одна из цепей ДНК соответственно А-формы, В-формы, Z-формы. На рисунках выделен и подписан шаг спирали в ангстремах.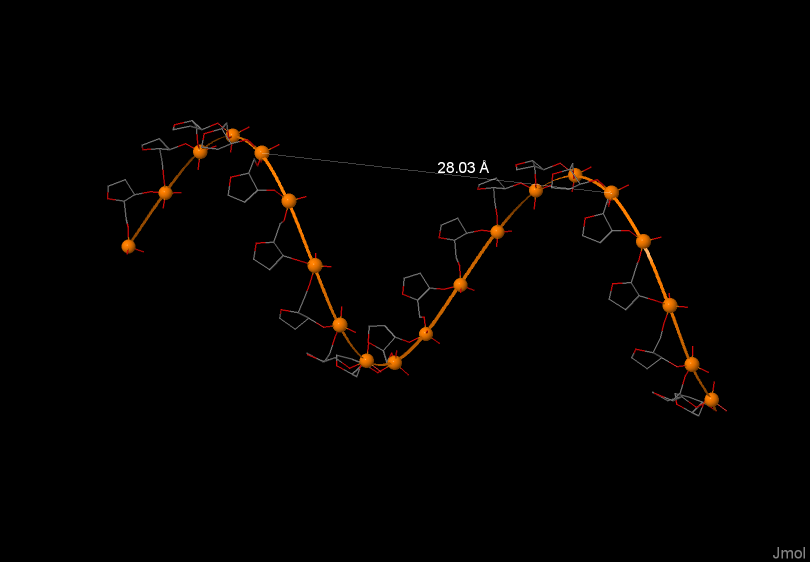
|
A-форма |
B-форма |
*Z-форма |
Тип спирали (правая или левая) |
Правая |
Правая |
Левая |
Шаг спирали (A) |
28.03 |
33.75 |
43.5 |
Число оснований на виток |
11 |
10 |
12 |
Ширина большой бороздки |
16.81 |
17.91 |
18.30 |
Ширина малой бороздки |
7.98 |
11.69 |
8.68 |
Малая бороздка
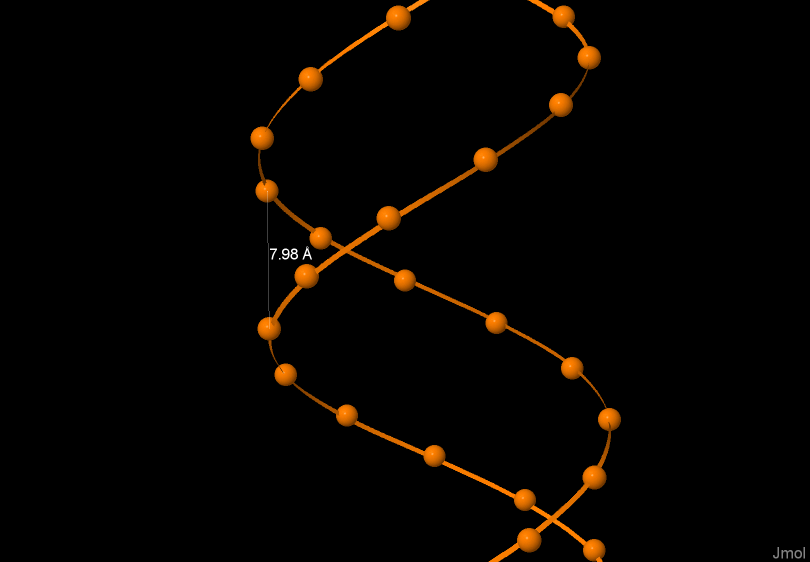
Большая бороздка
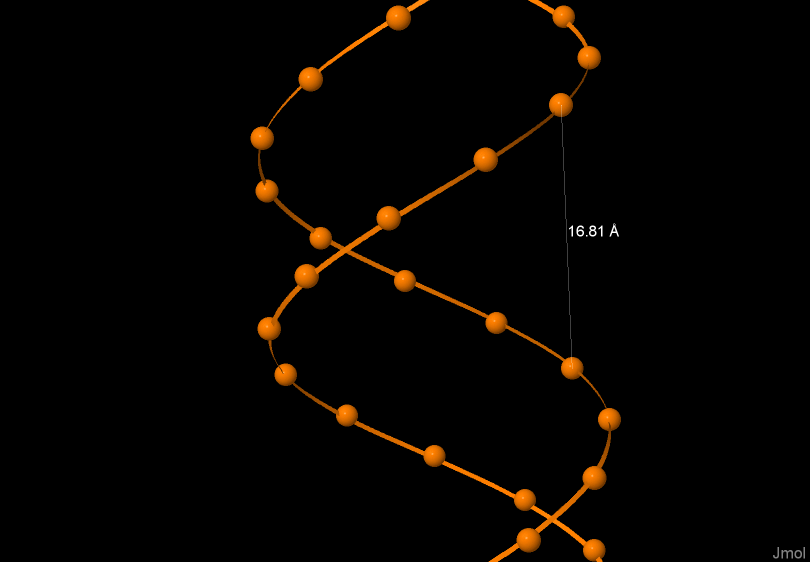
Малая бороздка
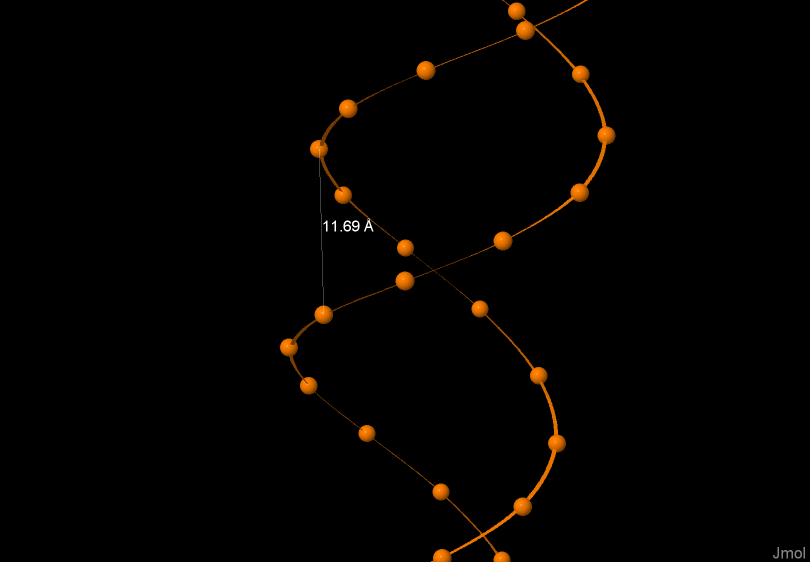
Большая бороздка
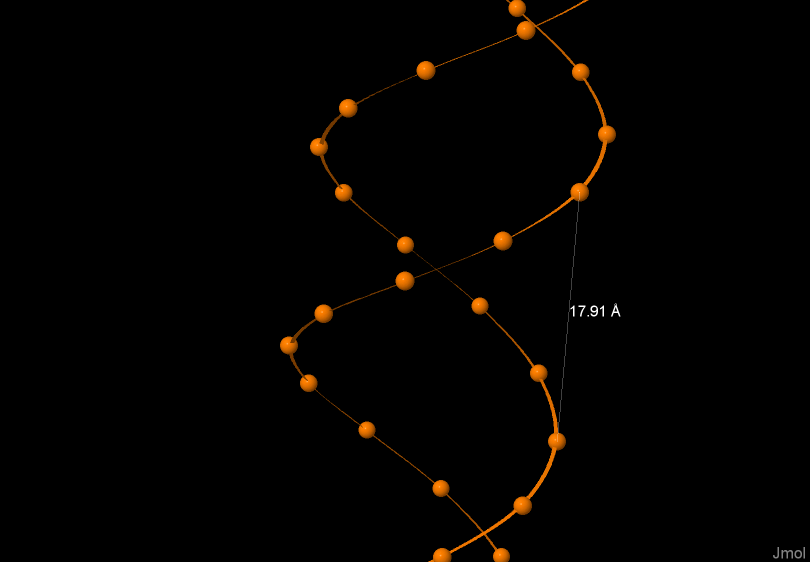
Малая бороздка
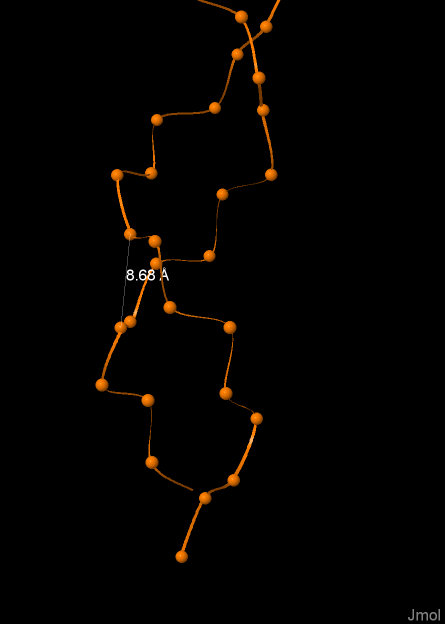
Большая бороздка
Сравнение торсионных углов в структурах А- и В-форм
С помощью команды Measurements->Click for torsion(dihedral) measurement в JMol измерим торсионные углы тимина в А- и В-форме. Эта команда позволяет определять торсинные углы заданной последовательности из четырех последовательно соединенных атомов.
| α (P - O5') | β (O5' - C5') | γ (C5' - C4') | δ (C4' - C3') | ε (C3' - O3') | ζ (O3' - P) | χ (C1' - N) | |
| A-ДНК | -51.70 | 174.80 | 41.70 | 79.1 | -147.80 | -75.20 | -157.20 |
| B-ДНК | -29.89 | 136.32 | 31.17 | 143.34 | -140.8 | -160.50 | -97.97 |
| Значения торсионных углов из презентации | |||||||
| A-ДНК | -62 | 173 | 52 | 88 или 3 | 178 | -50 | -160 |
| B-ДНК | -63 | 171 | 54 | 123 или 131 | 155 | -90 | -117 |
Определение параметров структур нуклеиновых кислот с помощью программ пакета 3DNA
Определение торсионных углов нуклеотидов
Определим торсионные углы в структуре 1ddn ДНК. Для это возьмем pdb-файл только с нуклеиновой кислотой, который мы
получили ранее: 1DNN_DNA.txt и переведем его в старый формат PDB, так как пакет 3DNA пока работает
только с ним.
Мы получим файл 1DDN_old.pdb. Программа find_pair определяет спаренные основания и положения спиралей в структуре. Полученные данные необходимы для работы программы analyze. Можно перенаправить результат работы find_pair на вход программе analyze:
В результате мы получим ряд файлов с описанием разных параметров структуры, в файле 1DDN_old.out
можно найти описание водородных связей, значения всех торсионных углов, ширину малой и большой бороздки.
Для удобства, значения торсионных углов рассматриваемой структуры приведены в файле
1ddn_torsion_angles.txt.
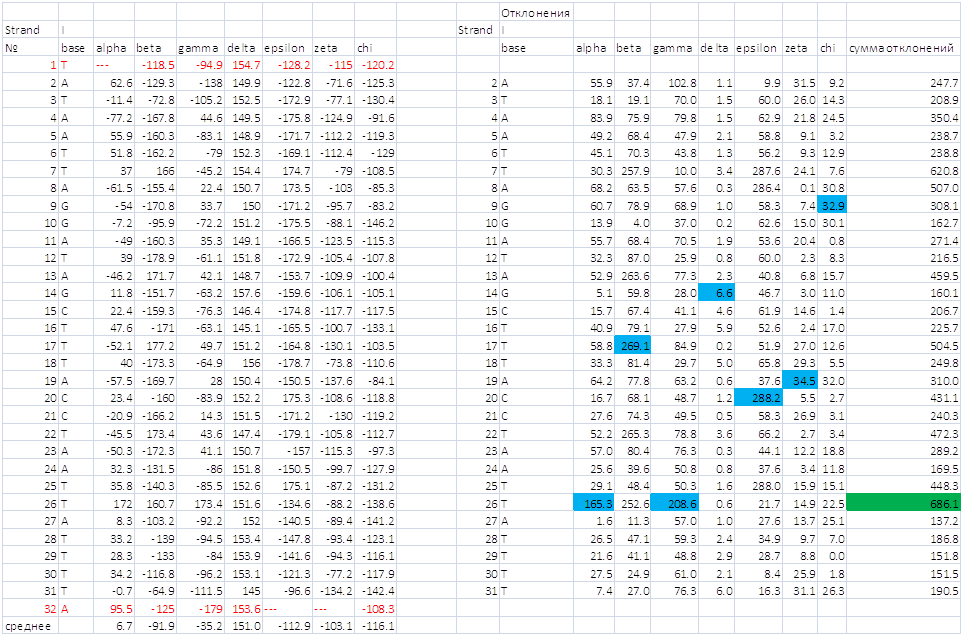
Теперь с помощью Excel определим среднее значение каждого из торсионных углов, краевые нуклеотиды при этом не рассматриваем. Определим номер самого "деформированного" нуклеотида (с наиболее отклоняющимся значением какого-либо из торсионных углов или нескольких углов). Получим файл strands.xls
Синим цветом выделены углы тех оснований первой цепи, для которых по данному углу наблюдается наибольшее отклонение.
Красным цветом выделены краевые нуклеотиды.
Синим цветом выделены углы тех оснований второй цепи, для которых по данному углу наблюдается наибольшее отклонение.
Красным цветом выделены краевые нуклеотиды.
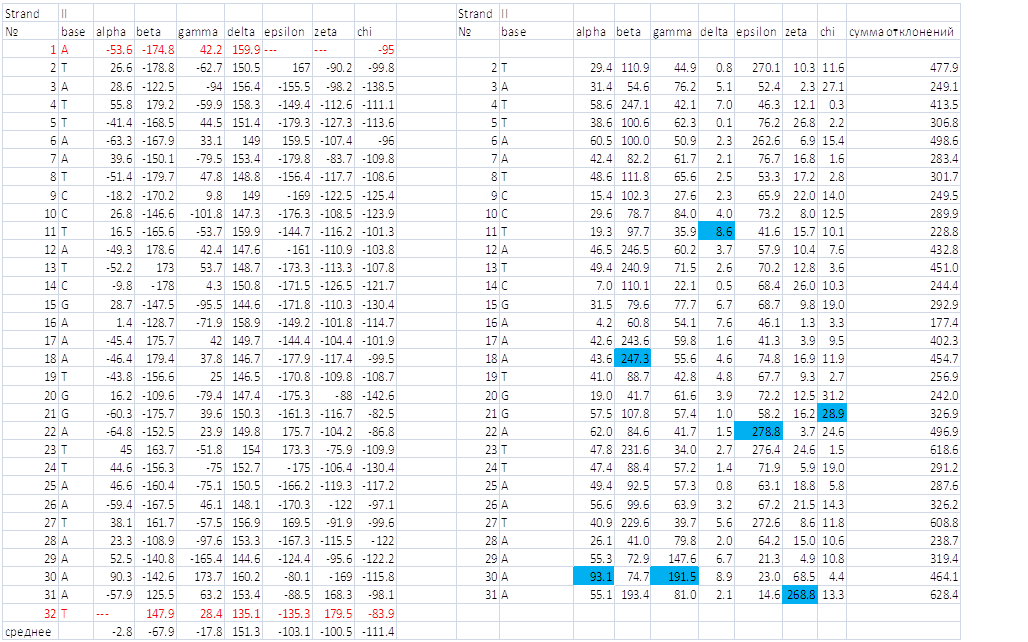
Из рис 1. и рис.2 наглядно видно, что если рассматривать отклонение только в одном угле, то однозначно самый деформированный
нуклеотид - С20 из первой цепи. Отклонение в угле ε - 288,2. Если рассматривать самый деформированный нуклеотид,
как тот, у кого наибольшая сумма отклонений,то это T26 из первой цепи (сумма отклонений этого нуклеотида выделена зеленым
на рис.1).
Определение структуры водородных связей
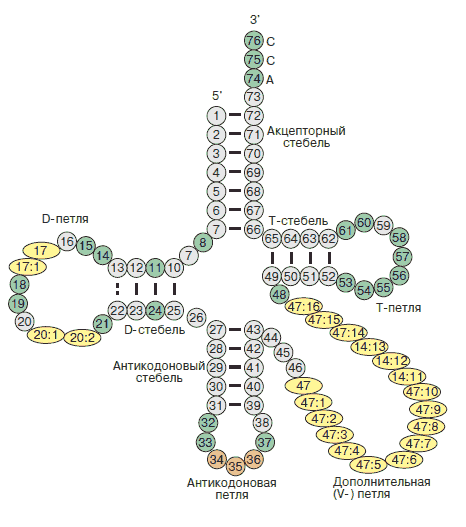
Определим номера нуклеотидов, образующих стебли(stems) во вторичной структуры 1G59 тРНК. Для этого откроем файл 1G59_RNA_old.out, в нем найдем информацию о спаренных основаниях, запишем ее в отдельный файл 1G59_pairs.txt.
- Акцепторный стебель состоит из участка 501-507 и комплементарного ему участка 566-572.
- T-стебель состоит из участка 549-553 и комплементарного ему участка 561-565.
- D-стебель состоит из участка 510-512 и комплементарного ему участка 523-525.
- Антикодоновый стебель состоит из участка 538-544 и комплементарного ему участка 526-532.
Синим цветом выделен акцепторный стебель.
Желтым - T - стебель
Красным - Антикодоновый стебель
Зеленым - D-стебел
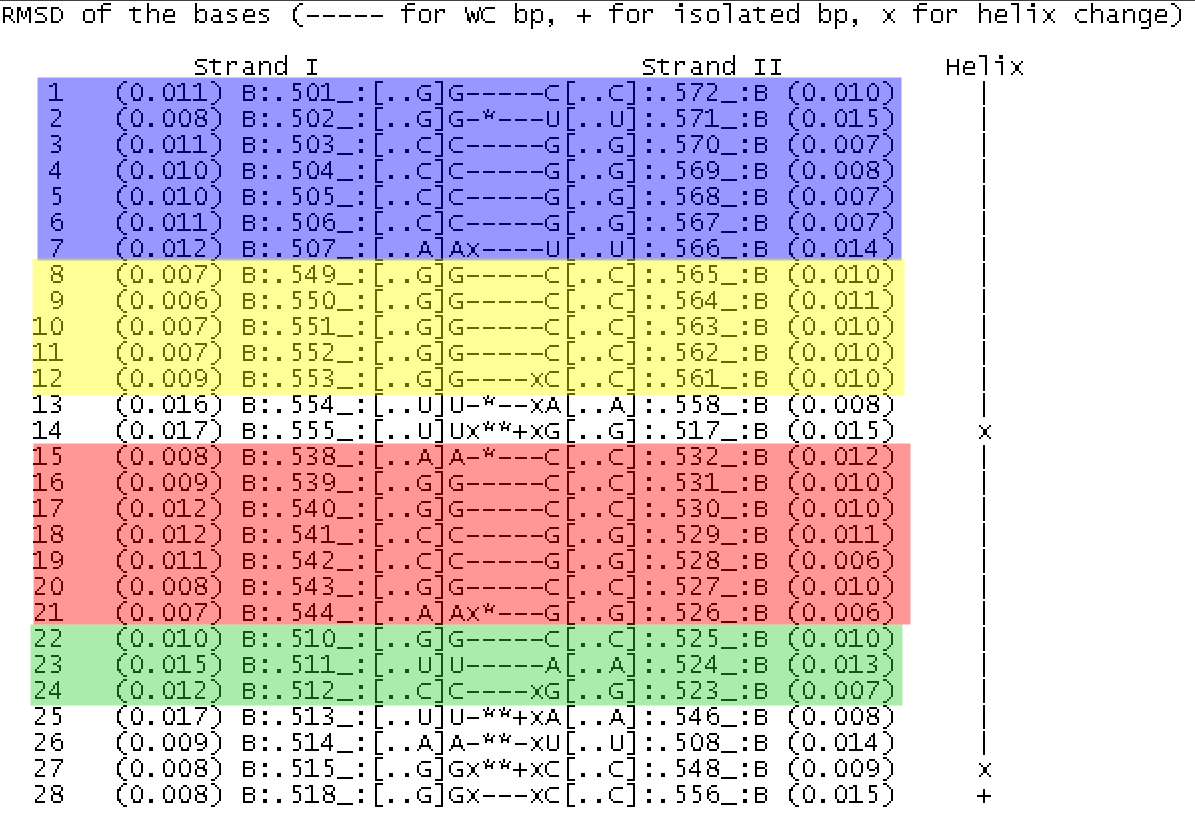
Синим цветом выделен акцепторный стебель.
Желтым - T - стебель
Красным - Антикодоновый стебель
Зеленым - D-стебел
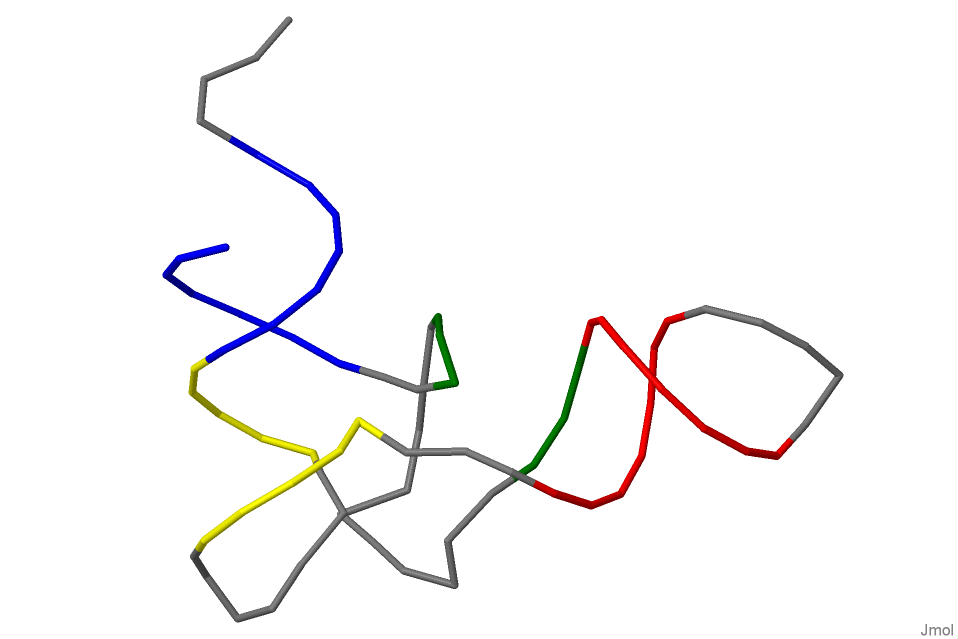
Скрипт, при помощи которого было получено изображение script.txt
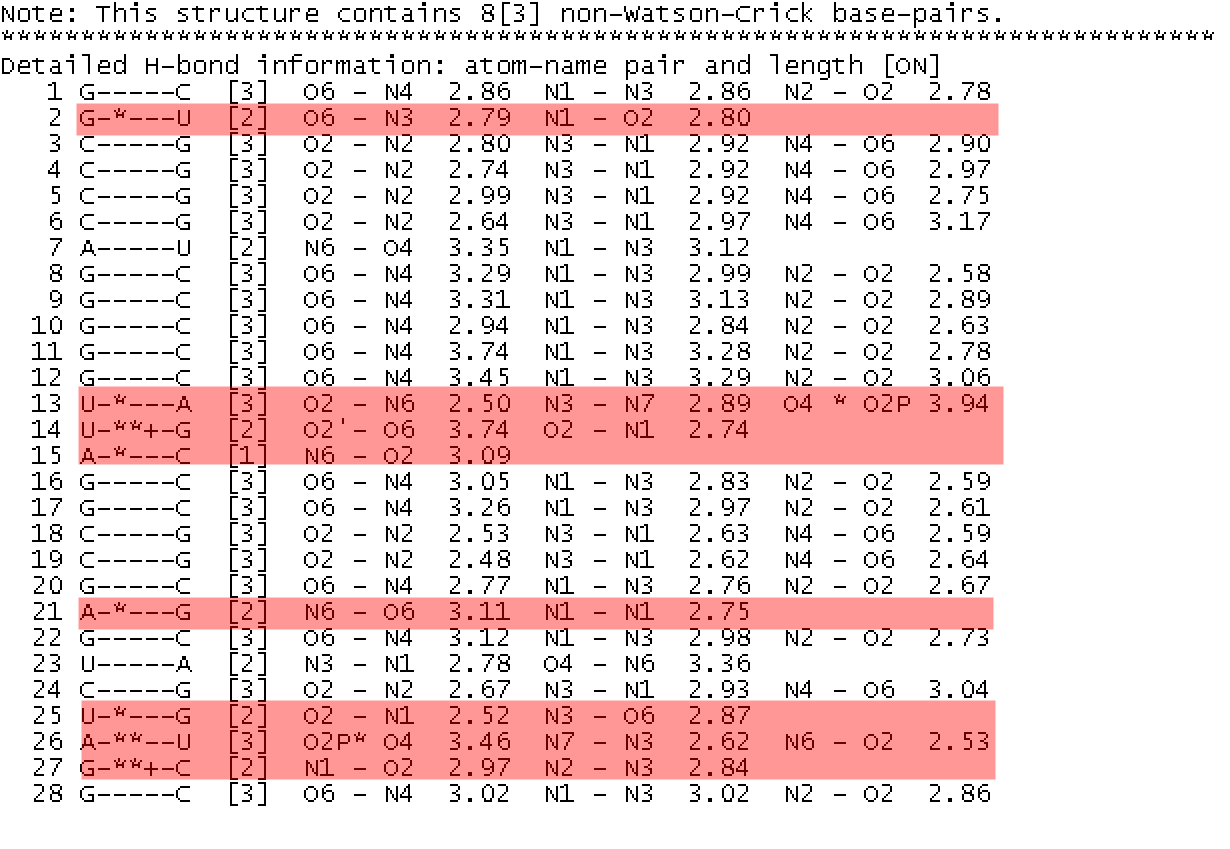
Теперь определим неканонические пары оснований в структуре тРНК. Для этого обратимся к тому же файлу
1G59_RNA_old.out, найдем в нем нужную информацию и поместим в отдельный файл
noncanonic.txt.
Неканонические пары отмечены звездочкой (8 штук), причем такие пары могут быть принципиально
двух разных типов: некомплементарные пары(то есть не A-U и G-C) и комплементарные пары между которыми есть неканонические водородные
связи. На рис.4 выделены красным цветом неканонические пары структуры 1G59 тРНК.
Неканоничсекие пары выделены красным цветом.
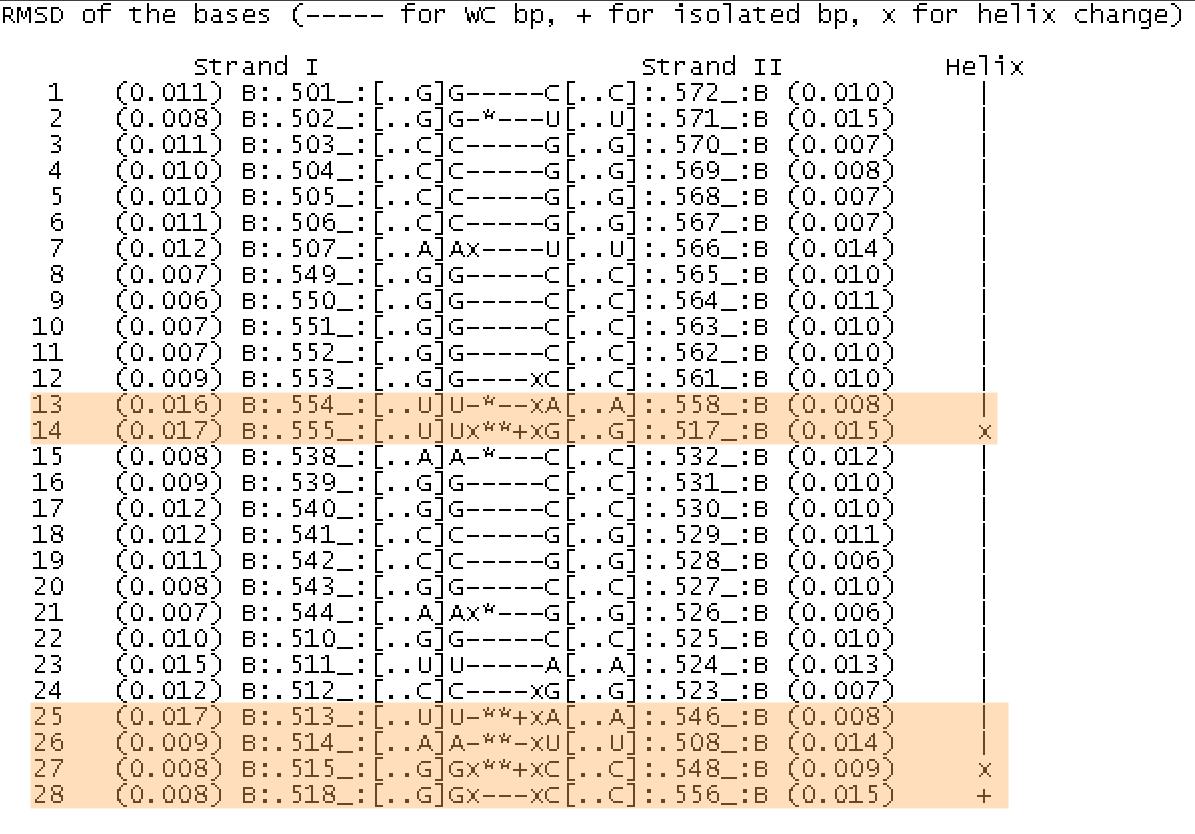
Определим, есть ли дополнительные водородные связи в тРНК, стабилизирующие ее третичную структуру (для этого следует рассмотреть
комплементарные пары, не имеющие отношения к стеблям).
Пары выделены оранжевым цветом.
Нахождение возможных стекинг-взаимодействий
Получим нужные нам файлы с информацией о структуре тРНК командой :
Откроем файл 1G59_old.out с характеристикой структуры 1G59 тРНК. В нем можно найти информацию
о последовательном перекрывании пар оснований, для удобство поместим ее в отдельный файл stecking.txt
Чем больше пары перекрываются, тем сильнее stacking.
Из файла видим, что 2ая пара имеет максимальное перекрываение - 13.51 квадратных ангстрем.
Информацию о пространственной структуре интересующей нас пары можно получить из файла stacking.pdb,
который генерируется программой find_pair, использованной нами ранее. В этом файле есть pdb модели всех перекрывающихся попарно пар.
Вычленим координаты нужной нам пары в файл pair.pdb.
Теперь визуализируем данную пару в Jmol.
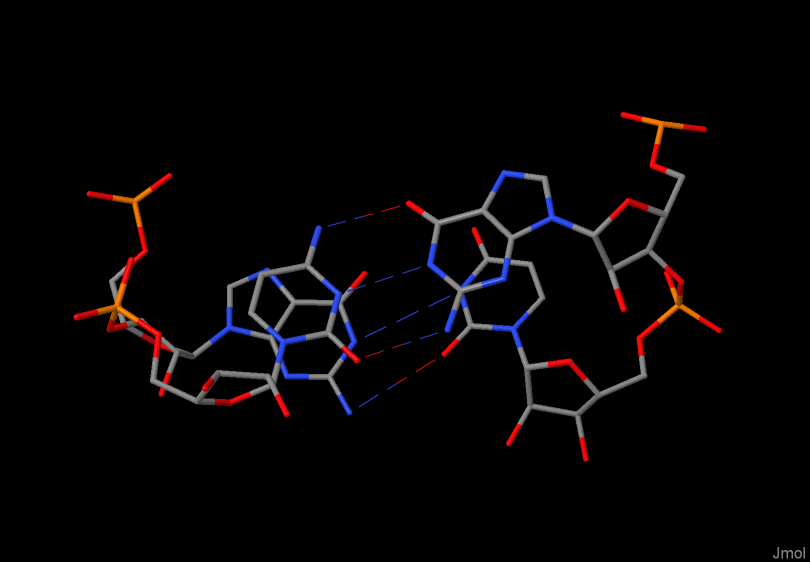
Аналогичную визуализацию можно проделать и с помощью программ:
ex_str -N stacking.pdb stepN.pdb
Где N - номер блока с нужной парой.
Полученый pdb-файл можно конвертировать в формат ps:
stack2img -cdolt stepN.pdb stepN.ps
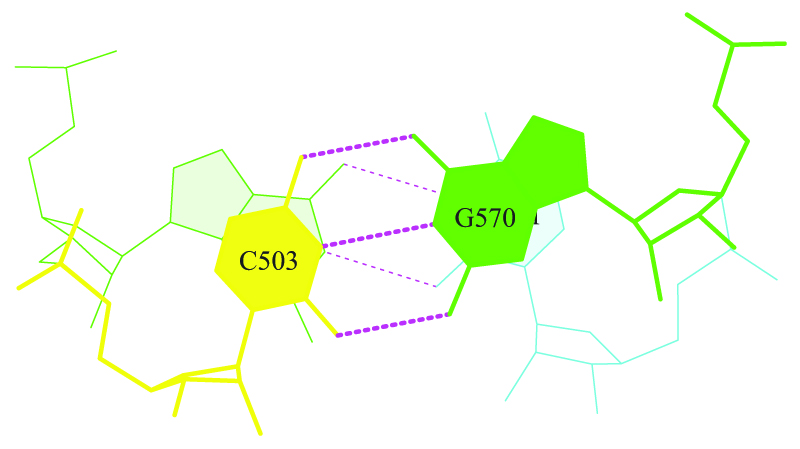
.