1. Сравнение выравнивания одних и тех же последовательностей тремя разными программами MSA
Для сравнения результатов работы трёх программ множественного выравнивания был выбран ГТФазный домен SRP54-подобных белков, участвующих в распознавании сигнальной последовательности в синтезируемом белке, связывании с рибосомой, замедляя трансляцию, доставке рибосомы и белка к мембране ЭР или плазматической мембране у прокариот. Гидролиз GTP обеспечивает энергию для диссоциации комплекса SRP-рибосома-белок.
Выравнивание было произведено при помощи трех программ: Mafft, Muscle, ClustalO, причем в качестве референсного было выбрано выравнивание Mafft. Результаты сравнения выравниваний с помощью программы MACHO представлены в таблицах 1 и 2 (указаны координаты колонок, выровненных одинаково).
| Block | Alignment_Mafft | Alignment_Muscle |
|---|---|---|
| 1 | 1-11 | 1-11 |
| 2 | 15-24 | 15-24 |
| 3 | 35-64 | 35-64 |
| 4 | 99-126 | 100-127 |
| 5 | 162-165 | 160-163 |
| 6 | 168-218 | 166-216 |
| Block | Alignment_Mafft | Alignment_ClustalO |
|---|---|---|
| 1 | 1-12 | 1-12 |
| 2 | 19-24 | 19-24 |
| 3 | 33-58 | 33-58 |
| 4 | 60-61 | 60-61 |
| 5 | 63-70 | 63-70 |
| 6 | 99-125 | 97-123 |
| 7 | 128-132 | 126-130 |
| 9 | 162-164 | 157-159 |
| 10 | 168-223 | 163-218 |
Обсуждение результатов
Для пары выравниваний Mafft/Muscle длина последовательностей и процент совпадающий колонок соответственно равны: 223/221 и 60.54%/61.09 %. Для пары выравниваний Mafft/ClustalO: 238/218 и 65.47%/66.97%
Из сравнения следует, что большим сходством (числом одинаково выровненных колонок) обладают программы Mafft и ClustalO, но при этом наблюдается большая разница в длине.
Примечательно, что в выравнивании Muscle индели иногда идут через через 1-2 нуклеотида (рис. 1), что с эволюционной точки зрения очень маловероятно, а программа ClustalO делает более "компактное" выравнивание, по сравнению с Mafft, кроме того, алгоритм ClustalO более похож на Muscle, чем на Mafft (с оценкой в более 68% совпадающих колонок).

2. Построение выравнивания по совмещению структур и его сравнение
Для этого задания из базы Pfam были выбраны 3D структуры и последовательности трёх белков, содержащих 2Fe-2S железосерный связывающий домен (PF00111): SPINACH FERREDOXIN from Spinacia oleracea (1A70), [2Fe-2S] Domain of Methane Monooxygenase Reductase from Methylococcus capsulatus (1JQ4), PUTIDAREDOXIN from Pseudomonas putida (1PDX). Для всех белков взята цепь A, получено совмещение пространственных структур (рис.2) и построено два выравнивания: ручное из совмещения структур и автоматическое программой Mafft.
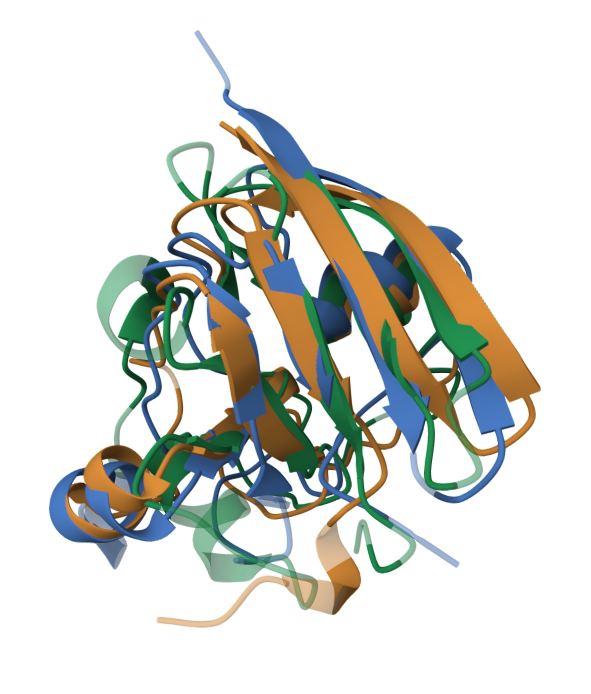
Для пары выравниваний Manual/Mafft длина последовательностей и процент совпадающий колонок соответственно равны: 121/116 и 39.67% /41.38%. Такой относительно небольшой процент совпадения (табл. 3) можно объяснить несовершенностью ручного выравнивания за счёт большого количества ненужных гэпов. Тем не менее, хорошо выравниваются в обоих способах участки 6-11 и 18-21, 28-32, что соответствует совместившимся бета-листам и альфа-спиралям в структурах белков, поэтому можно сделать вывод, что пространственные структуры трёх белков сходны, за исключением петель - участков, не образующих регулярные вторичные структуры, так как они в меньшей степени влияют на геометрию белка, а значит, могут быть более функционально гибкими и вариабельными.
| Block | Alignment_Manual | Alignment_Mafft |
|---|---|---|
| 1 | 1-11 | 1-11 |
| 2 | 17-37 | 16-36 |
| 3 | 42-44 | 41-43 |
| 4 | 53-62 | 51-60 |
| 5 | 75-75 | 69-69 |
| 6 | 88-89 | 87-88 |
Описание MAFFT - программы MSA
Множественное выравнивание последовательностей — это фундаментальная задача в биоинформатике, используемая для анализа структурных, функциональных и эволюционных связей. Традиционные методы, такие как Clustal, MUSCLE, T-COFFEE, хотя и достаточно точны, сталкиваются с проблемами масштабируемости и скорости при обработке огромных данных, таких как геномные и метагеномные последовательности. Эту проблему решает программа MAFFT, за счёт следующих принципов работы:
1. Использование FFT для быстрого поиска гомологичных сегментов
Преобразование Фурье (FFT) применяется для быстрого вычисления корреляционных функций между последовательностями. Для аминокислот используют векторы, компоненты которых — это значения физических свойств, например, объема v(a) и полярности p(a), нормализованные по среднему и стандартному отклонению.
v̂(a) = [v(a) – v̄]/σn (1) и p̂(a) = [ p(a)– p̄]/σp
В классических методах, таких как Needleman–Wunsch или Smith–Waterman, сравнение двух последовательностей — это (O(N^2)) операция. В случае больших наборов данных это становится непрактично. В MAFFT используют FFT, чтобы быстро находить похожие участки, что позволяет снизить сложность до порядка (O(N \log N)).
2. После выявления гомологичных участков с помощью FFT используют прогрессивное выравнивание, основанное на матрице сходства.
3. Итеративное рафинирование — для повышения точности итогового выравнивания. Прогрессивные методы выравнивают последовательности последовательно, начиная с наиболее близких пар, постепенно добавляя более отдалённые, но ошибки, допущенные на ранних этапах, не исправляются. Основные принципы итеративного уточнения в MAFFT: разбиение исходного выравнивания на подгруппы, повторное выравнивание подгрупп, замена повторно выровненными участками соответствующих участков в общем множественном выравнивании, многократное повторение процесса (обычно несколько десятков или сотен итераций) до наблюдения улучшения оценки.
4. В отличие от программы ClustalW, система оценки MAFFT проста: матрица сходства фиксирована для любых входных данных, и даже штраф за расширение пропуска не включен явно в алгоритм динамического программирования. Тем не менее, точность NW-NS-2/FFT-NS-2 сопоставима с точностью CLUSTALW.
Тесты на симуляциях и реальных данных
В рамках экспериментов использовались симуляции, созданные с помощью программы ROSE, моделирующей разные уровни гомологии. В сравнении с классическими методами, такими как ClustalW, MUSCLE, T-COFFEE, и новыми — FFT-NS-2 и FFT-NS-i, новые алгоритмы показывают: высокую скорость, особенно при больших объёмах данных; уменьшение времени работы с увеличением числа последовательностей; сравнимую или превосходящую точность, особенно при выравнивании далёких последовательностей.